The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
![]()
![]()
![]()
This package is comprised of functions that facilitate the identification of areas of importance for biodiversity, such as Key Biodiversity Areas (KBAs), based on individual tracking data. For further detail concerning the method itself, please refer to this paper by Beal et al. (2021).
Key functions include utilities to estimate individual core use areas, the level of representativeness of the tracked sample, and overlay individual distributions to identify important sites at the population level. Other functions assist in plotting the results, formatting your data set, and splitting and summarizing individual foraging trips.
You can download the stable version from CRAN with:
install.packages("track2KBA")Or you can download the development version from GitHub with:
install.packages("devtools", dependencies = TRUE)
devtools::install_github("BirdLifeInternational/track2kba", dependencies=TRUE) # development version - add argument 'build_vignettes = FALSE' to speed it up Now we will use tracking data collected at a seabird breeding colony
to illustrate a track2KBA workflow for identifying
important sites. It is important to note that the specific workflow you
use (i.e., which functions and in what order) will depend on the species
of interest and the associated data at hand.
First, in order for the data to work in track2KBA
functions, we can use the formatFields function to format
the important data columns needed for analysis. These are: a DateTime
field, Latitude and Longitude fields, and an ID field (i.e. individual
animal, track, or trip).
library(track2KBA) # load package
data(boobies)
# ?boobies # for some background info on the example data set
dataGroup <- formatFields(
dataGroup = boobies,
fieldID = "track_id",
fieldDate = "date_gmt",
fieldTime = "time_gmt",
fieldLon = "longitude",
fieldLat = "latitude"
)
str(dataGroup)If your data come from a central-place foraging species (i.e. one
which makes trips out from a centrally-located place, such as a nest in
the case of a bird), you can use tripSplit to split up the
data into discrete trips.
In order to do this, you must identify the location(s) of the central place(s) (e.g. colony-center, or nest sites).
library(dplyr)
# here we know that the first points in the data set are from the colony center
colony <- dataGroup %>%
summarise(
Longitude = first(Longitude),
Latitude = first(Latitude)
)Our colony dataframe tells us where trips originate from. Then we can set some parameters to decide what constitutes a trip. To do that we should use our understanding of the movement ecology of the study species. In this case we know our seabird travels out to sea on the scale of tens of kilometers, so we set innerBuff (the minimum distance from the colony) to 3 km, and duration (minimum trip duration) to 1 hour. returnBuff can be set further out in order to catch incomplete trips, where the animal began returning, but perhaps due to device failure the full trip wasn’t captured.
Optionally, we can set rmNonTrip to TRUE which will remove
the periods when the animals were not on trips. The results of
tripSplit can be plotted using mapTrips to see
some examples of trips.
str(dataGroup)
trips <- tripSplit(
dataGroup = dataGroup,
colony = colony,
innerBuff = 3, # kilometers
returnBuff = 10,
duration = 1, # hours
rmNonTrip = TRUE
)
mapTrips(trips = trips, colony = colony)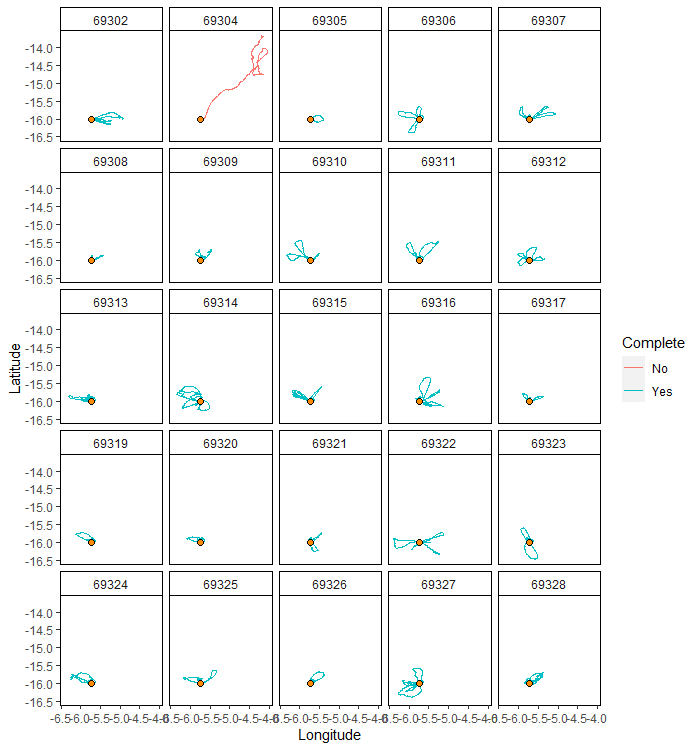
Then we can summarize the trip movements, using
tripSummary. First, we can filter out data from trips that
did not return to the vicinity of the colony (i.e. within
returnBuff), so they don’t skew the estimates.
trips <- subset(trips, trips$Returns == "Yes" )
sumTrips <- tripSummary(trips = trips, colony = colony)
sumTripsNow that we have an idea how the animals are moving, we can start with the process of estimating their space use areas, and identifying potentially important sites for the population!
track2KBA uses Kernel Density Estimation (KDE) to
produce space use estimates for each individual track. In order for
these to be accurate, we need to transform the tracking data to an
equal-area projection. We can use the convenience function
projectTracks to perform this projection. We can select
between an azimuthal or cylindrical projection, and decide whether to
center the projection on the data itself. Custom-centering is generally
a good idea for quick analyses as this will minimize distortion, however
it is important to remember that the resulting projection will be data
specific. So if you remove even one track and re-analyze, the projection
will differ between datasets. For formal analysis, the best solution is
to find a standard projection that is appropriate for your study
region.
tracks <- projectTracks( dataGroup = trips, projType = 'azim', custom=TRUE )
class(tracks)findScale provides options for setting the all-important
smoothing parameter in the KDE. This parameter decisions is of the
utmost importance, as it determines the scale at which the tracking
locations will be ‘smoothed’ into an estimate of the probability of use
of space by the animal. findScale calculates candidate
smoothing parameter values using several different methods.
If we know our animal uses an area-restricted search (ARS) strategy
to locate prey, then we can set the scaleARS=TRUE. This
uses First Passage Time analysis to identify the spatial scale at which
area-restricted search is occuring, which may then be used as the
smoothing parameter value.
hVals <- findScale(
tracks = tracks,
scaleARS = TRUE,
sumTrips = sumTrips)
hValsThe other values provided by findScale are more
simplistic methods of calculating the smoothing parameter.
href is the canonical reference method, and relates to the
number of points in the data and their spatial variance.
mag is the log of the average foraging range
(med_max_dist in the sumTrips output); this
methods only works for central-place foragers.
Next, we must select a smoothing parameter value. To inform our
decision, we ought to use our understanding of the species’ movement
ecology to guide our decision about what scale make sense. That is, from
the findScale output, we want to avoid using values which
may under- or over-represent the area used by the animals while
foraging.
Once we have chosen a smoothing value, we can produce KDEs for each
individual, using estSpaceUse. By default this function
isolates the core range of each track (i.e. the 50% utilization
distribution, or where the animal spends about half of its time) which
is a commonly used standard (Lascelles et al. 2016). However, another
quantile can be chose using the levelUD argument, or the
full utilization distritbution can be returned using
polyOut=FALSE.
The resulting KDEs can be plotted using mapKDE, which if
polyOut=TRUE shows each tracks’s core range in a different
color.
Note: here we might want to remove the trip start and end
points that fall within the innerBuff (i.e. 3 km) we set in
tripSplit, so that they don’t skew the at-sea distribution
towards to colony.
tracks <- tracks[tracks$ColDist > 3, ] # remove trip start and end points near colony
KDE <- estSpaceUse(
tracks = tracks,
scale = hVals$mag,
levelUD = 50,
polyOut = TRUE
)
mapKDE(KDE = KDE$UDPolygons, colony = colony)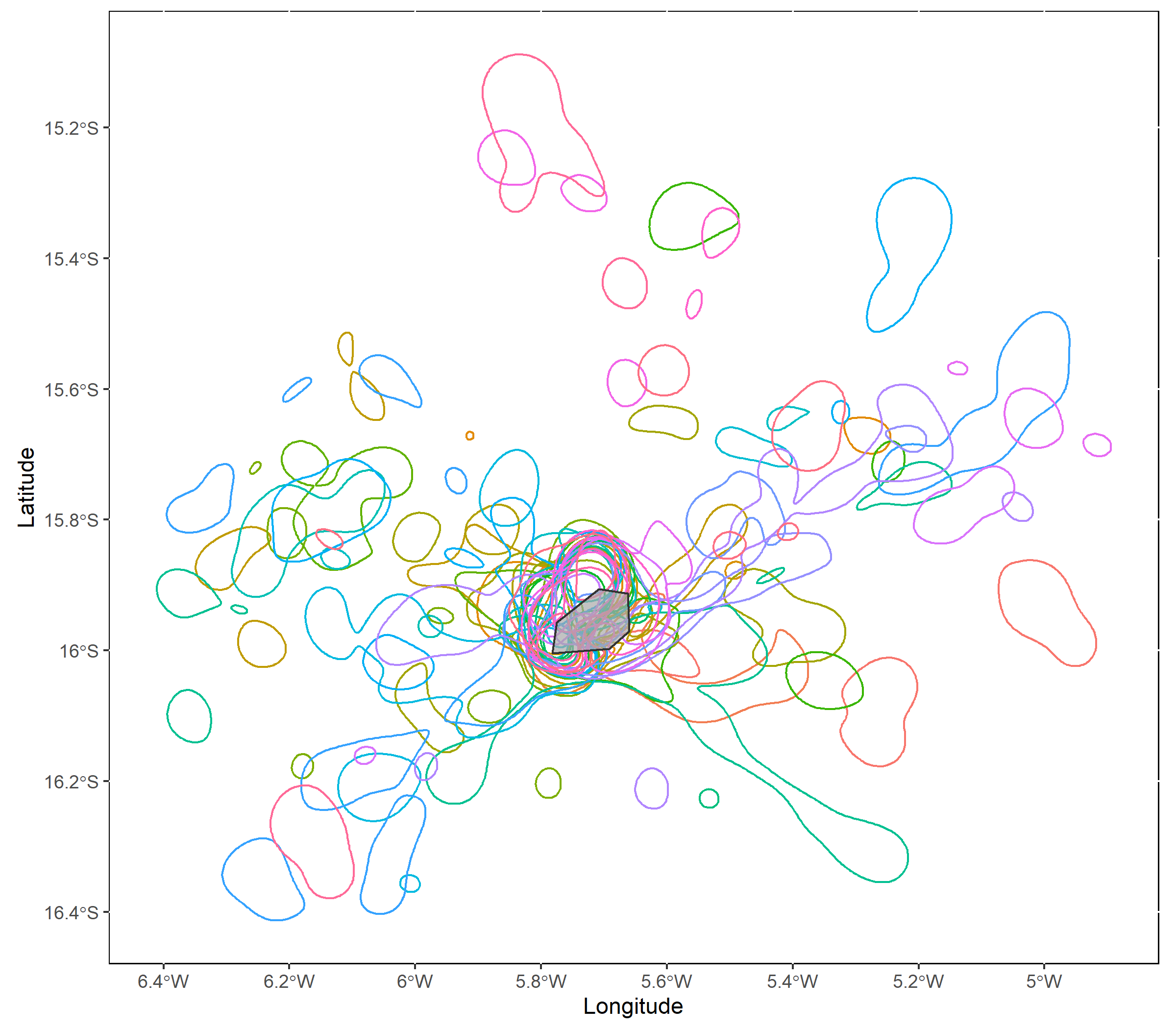 At this step we should verify that the smoothing parameter value we
selected is producing reasonable space use estimates, given what we know
about our study animals. Are the core areas much larger than expected?
Much smaller? If so, consider using a different value for the
At this step we should verify that the smoothing parameter value we
selected is producing reasonable space use estimates, given what we know
about our study animals. Are the core areas much larger than expected?
Much smaller? If so, consider using a different value for the
scale parameter.
The next step is to estimate how representative this sample of
animals is of the population. That is, how well does the variation in
space use of this sample of tracks encapsulate variation in the wider
population? To do this we can use the repAssess function.
This function repeatedly samples a subset of track core ranges, averages
them together, and quantifies how many points from the unselected tracks
fall within this combined core range area. This process is run across
the range of the sample size, and iterated a chosen number of times.
To do this, we need to supply Utilization Distributions to
repAssess (e.g., the output of estSpaceUse)
and the tracking data. We can choose the number of times we want to
re-sample at each sample size by setting the iteration
argument. The higher the number the more confident we can be in the
results, but the longer it will take to compute.
repr <- repAssess(
tracks = tracks,
KDE = KDE$KDE.Surface,
levelUD = 50,
iteration = 1,
bootTable = FALSE)The output is a dataframe, with the estimated percentage of
representativeness given in the out column.
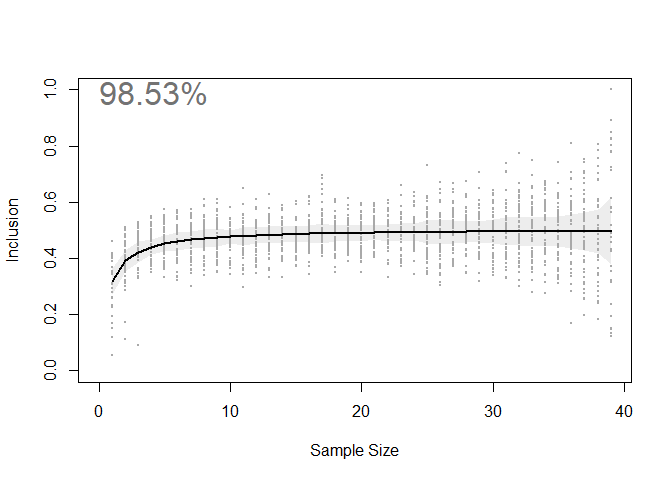
The relationship between sample size and the percent coverage of un-tested animals’ space use areas (i.e. Inclusion) is visualized in the output plot seen below.
By quantifying this relationship, we can estimate how close we are to an information asymptote. Put another way, we have estimated how much new space use information would be added by tracking more animals. In the case of this seabird dataset, we estimate that ~98% of the core areas used by this population are captured by the sample of 39 individuals. Highly representative!
Now, using findSite we can identify areas where animals
are overlapping in space and delineate sites that meet some criteria of
importance. Using the core area estimates of each individual track we
can calculate where they overlap. Then, we estimate the proportion of
the larger population in a given area by adjusting our overlap estimate
based on the degree of representativeness.
Here, if we have population size estimates, we can include this value
(using the popSize argument) to estimate the number of
individuals using a space, which can then use to compare against
population-level importance criteria (e.g. KBA criteria). If we don’t
have population size estimates to provide, findSite this
will output a proportion of the population instead.
If you desire polygon output of the overlap areas, instead of a
gridded surface, you can indicate this using the polyOut
argument.
Site <- findSite(
KDE = KDE$KDE.Surface,
represent = repr$out,
levelUD = 50,
popSize = 500, # 500 individual seabirds breed one the island
polyOut = TRUE
)
class(Site)If we specified polyOut=TRUE, then the output will be of
Simple Features class, which allows us to easily take advantage of the
ggplot2 plotting syntax to make an attractive map using
mapSite!
Sitemap <- mapSite(Site, colony = colony)
## in case you want to save the plot
# ggplot2::ggsave("KBAmap", device="png")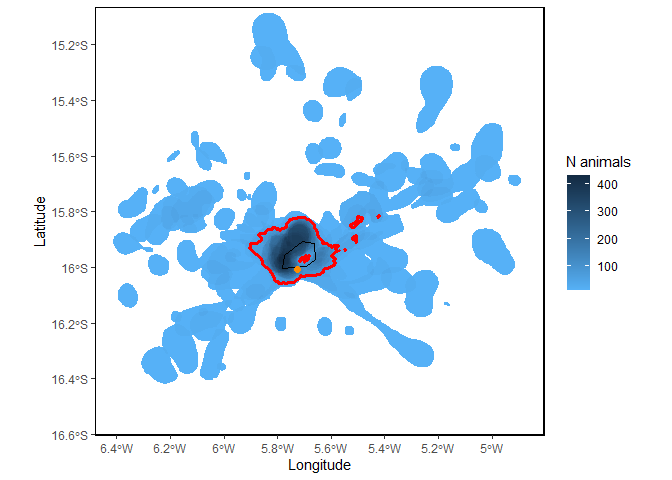
This map shows the number or proportion of individual animals in the population overlapping in space. The red lines indicate the ‘potential site’; that is, the areas used by a significant proportion of the local population, given the representativeness of the sample of tracked individuals. In this case, since representativeness is >90%, any area used by 10% or more of the population is considered important (see Lascelles et al. 2016 for details). The orange dot is the colony location and the black line is the coastline.
Note: it is possible to set the threshold of importance at the
population level yourself, using the thresh argument. Just
be aware that this is a crucial threshold parameter that will need
justifying if you are to eventually recommend a site for formal
acknowledgement as an important site for conservation.
Then, we can combine all the polygons within the ‘potentialSite’ area, and use, for example, the maximum number of individuals present in that area to assess whether it may merits identification as a Key Biodiversity Area according to the KBA standard.
potSite <- Site %>% dplyr::filter(.data$potentialSite==TRUE) %>%
summarise(
max_animals = max(na.omit(N_animals)), # maximum number of animals aggregating in the site
min_animals = min(na.omit(N_animals)) # minimum number using the site
)If in findSite we instead specify
polyOut=FALSE, our output will be a spatial grid of animal
densities, with each cell representing the estimated number, or
percentage of animals using that area. So this output is independent of
the representativness-based importance threshold.
mapSite(Site, colony = colony)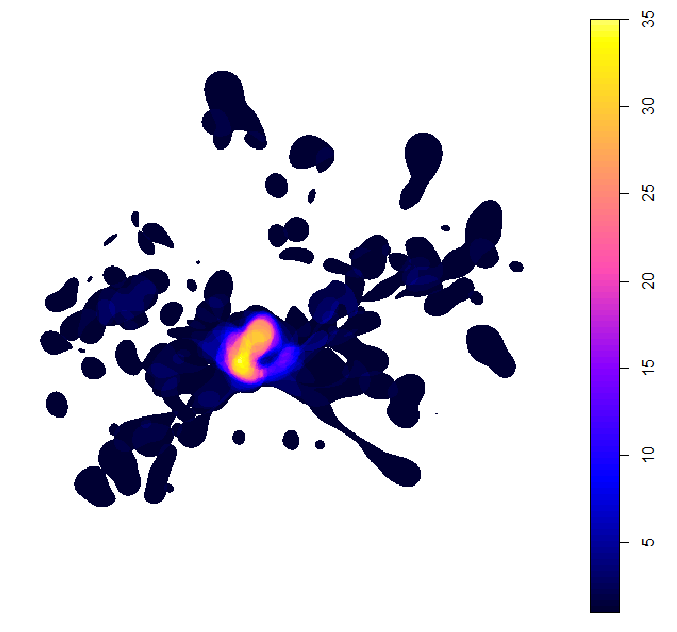
This plot shows the minimum estimated number of birds using the space around the breeding island.
If you use any functions in this package for your work, please use the following citation:
Beal, M., Oppel, S., Handley, J., Pearmain, E. J., Morera-Pujol, V., Carneiro, A. P. B., Davies, T. E., Phillips, R. A., Taylor, P. R., Miller, M. G. R., Franco, A. M. A., Catry, I., Patrício, A. R., Regalla, A., Staniland, I., Boyd, C., Catry, P., & Dias, M. P. (2021). track2KBA: An R package for identifying important sites for biodiversity from tracking data. Methods in Ecology and Evolution, 12(12), 2372-2378. https://doi.org/10.1111/2041-210X.13713
Oppel, S., Beard, A., Fox, D., Mackley, E., Leat, E., Henry, L., Clingham, E., Fowler, N., Sim, J., Sommerfeld, J., Weber, N., Weber, S., Bolton, M., 2015. Foraging distribution of a tropical seabird supports Ashmole’s hypothesis of population regulation. Behav Ecol Sociobiol 69, 915–926. https://doi.org/10.1007/s00265-015-1903-3
Lascelles, B. G., Taylor, P. R., Miller, M. G. R., Dias, M. P., Oppel, S., Torres, L., Hedd, A., Corre, M. L., Phillips, R. A., Shaffer, S. A., Weimerskirch, H., & Small, C. (2016). Applying global criteria to tracking data to define important areas for marine conservation. Diversity and Distributions, 22(4), 422–431. https://doi.org/10.1111/ddi.12411
Thanks to Annalea Beard for kindly sharing these example data for use in the package.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 766417.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.