The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
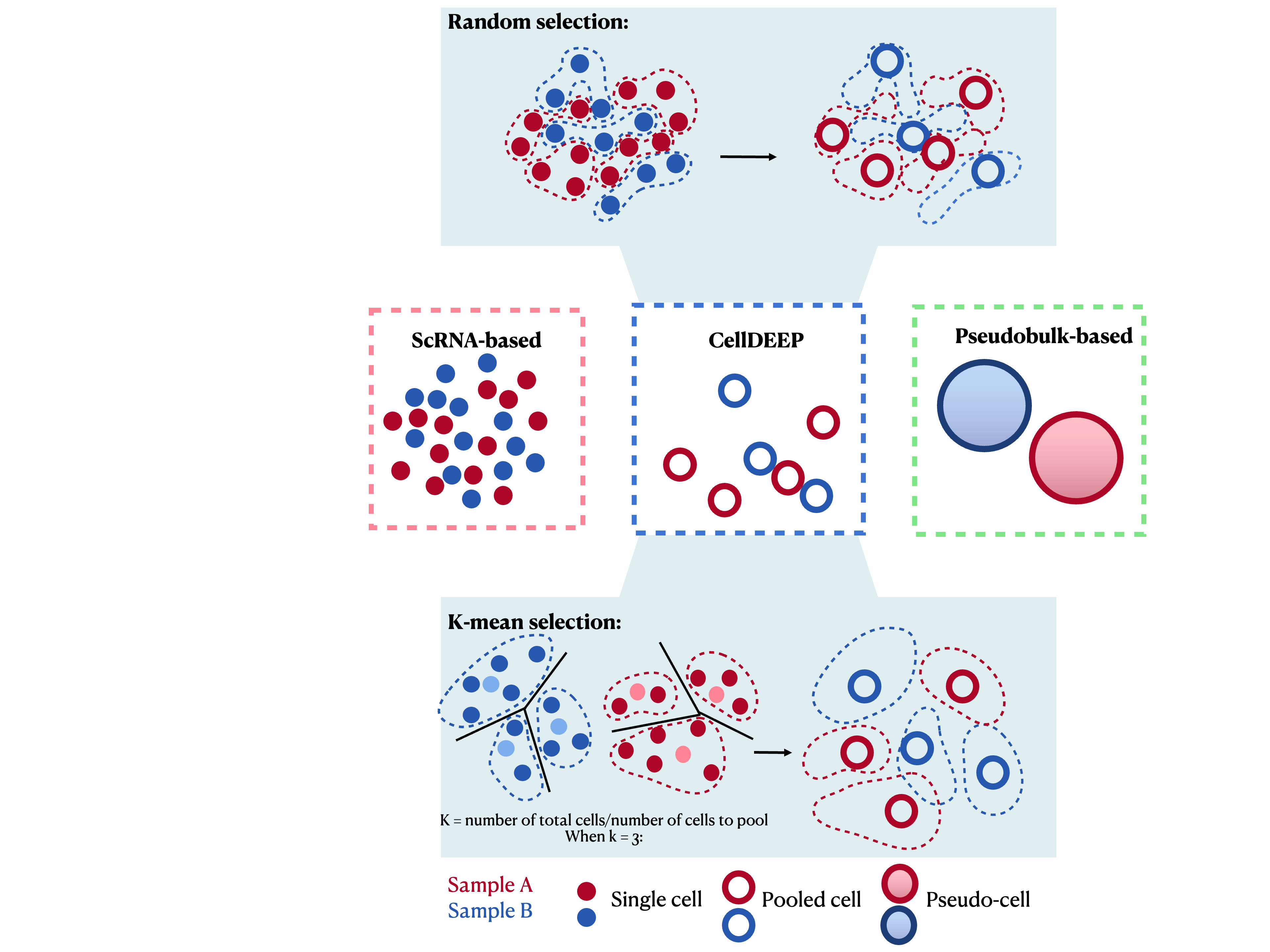
Single-cell RNA sequencing (scRNA-seq) allows us to explore gene expression at an unprecedented resolution, but it faces significant challenges like high dropout rates and data sparsity. CellDEEP was developed to bridge the gap between robust but coarse pseudobulk methods and sensitive but potentially biased single-cell methods.
Check our paper for details: https://www.biorxiv.org/content/10.64898/2026.03.09.710522v1
You can install the development version of CellDEEP via github:
devtools::install_github("sii-scRNA-Seq/CellDEEP")Before using, don’t forget library it:
library(CellDEEP)To quickly run CellDEEP, pass your metadata column names directly
into FindMarker.CellDEEP:
data("sim")
# Pool defaults to TRUE
de.test <- FindMarker.CellDEEP(sim,
group_id = "Status",
sample_id = "DonorID",
cluster_id = "cluster_id",
Pool = TRUE,
test.use = "wilcox",
n_cells = 3,
min_cells_per_subgroup = 1,
cell_selection = "random",
readcounts = "sum",
logfc.threshold = 0.25,
ident.1 = "Case",
ident.2 = "Control")This section introduce what is updated.
For version 1.0.1:
1. FindMarker.CellDEEP Pool default should be TRUE.
2. Not clear what is cell_cutoff, replaced with new parameter
3. Change “pool_way” to cell_selection
4. Vignette easy to access/read
5. Change toy data to simulated data, will generate DE result now.
For publish version(1.0.0):
Delete code used for experiment, keep only CellDEEP function code.
Delete all the comments, clean the code.
Rename pooling function as CellDEEP.Kmean and CellDEEP.Random.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.