The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
![]()
![]()
![]()
The DImodels package is designed to make fitting
Diversity-Interactions models easier. Diversity-Interactions (DI) models
(Kirwan et al 2009) are a set of tools for analysing and interpreting
data from experiments that explore the effects of species diversity
(from a pool of S species) on community-level responses. Data
suitable for DI models will include (at least) for each experimental
unit: a response recorded at a point in time, and a set of proportions
of S species \(`p_1`\), \(`p_2`\), …, \(`p_S`\) from a point in time prior to the
recording of the response. The proportions sum to 1 for each
experimental unit.
Main changes in the package from version 1.3.2 to version 1.3.3
contrast_matrix() function is introduced that creates
contrast matrices using species proportions which can be passed onto the
contrasts_DI() function to test for contrasts using a DI
model object.compare_communities() function is added which allows
for pairwise comparisons between various specific communities and
assigns compact letter displays (CLD) to each community.DImodel are
stacked onto the model object as attributes and can be accessed using
the attributes() and attr() functions.Main changes in the package from version 1.3.1 to version 1.3.2
predict() function now allows predicting the
response for species communities that do not sum to 1, but with a
warning notifying the user about this.Main changes in the package from version 1.3 to version 1.3.1
fortify() function method has been added to
supplement the data fitted to a linear model with model fit
statistics.describe_model() function is added which can be used
to get a short text summary of any DI model.attributes() function.Main changes in the package from version 1.2 to version 1.3
DI() and autoDI() functions now have
an additional parameter called ID which enables the user to
group the species identity effects (see examples below).predict() function now has flexibility to calculate
confidence and prediction intervals for the predicted values.Main changes in the package from version 1.1 to version 1.2
predict(): Make predictions from a fitted DI model
without having to worry about theta, and the interaction terms in the
data.contrasts_DI(): Create contrasts for a DI model.Main changes in the package from version 1.0 to version 1.1
DI_data_prepare() is now superseded by
DI_data() (see examples below)DImodels
installation and loadThe DImodels package is installed from CRAN and loaded
in the typical way.
install.packages("DImodels")
library("DImodels")It is recommended that users unfamiliar with Diversity-Interactions
(DI) models read the introduction to DImodels, before using
the package. Run the following code to access the documentation.
?DImodelsThere are seven example datasets included in the
DImodels package: Bell, sim1,
sim2, sim3, sim4,
sim5, Switzerland. Details about each of these
datasets is available in their associated help files, run this code, for
example:
?sim3In this vignette, we will describe the sim3 dataset and
show a worked analysis of it.
The sim3 dataset was simulated from a functional group
(FG) Diversity-Interactions model. There were nine species in the pool,
and it was assumed that species 1 to 5 come from functional group 1,
species 6 and 7 from functional group 2 and species 8 and 9 from
functional group 3, where species in the same functional group are
assumed to have similar traits. The following equation was used to
simulate the data.
y = \sum_{i=1}^{9}\beta_ip_i + \omega_{11}\sum_{\substack{i,j = 1 \\ i<j}}^5p_ip_j + \omega_{22}p_6p_7 + \omega_{33}p_8p_9 \\ + \omega_{12}\sum_{\substack{i \in {1,2,3,4,5} \\ j \in {6,7}}}p_ip_j + \omega_{13}\sum_{\substack{i \in {1,2,3,4,5} \\ j \in {8,9}}}p_ip_j + \omega_{23}\sum_{\substack{i \in {6,7} \\ j \in {8,9}}}p_ip_j + \gamma_k + \epsilonWhere \(`\gamma_k`\) is a treatment effect with two levels (k = 1,2) and \(`\epsilon`\) was assumed IID N(0, \(`\sigma^2`\)). The parameter values are in the following table.
| Parameter | Value | Parameter | Value | |
|---|---|---|---|---|
| \(`\beta_1`\) | 10 | \(`\omega_{11}`\) | 2 | |
| \(`\beta_2`\) | 9 | \(`\omega_{22}`\) | 3 | |
| \(`\beta_3`\) | 8 | \(`\omega_{33}`\) | 1 | |
| \(`\beta_4`\) | 7 | \(`\omega_{12}`\) | 4 | |
| \(`\beta_5`\) | 11 | \(`\omega_{13}`\) | 9 | |
| \(`\beta_6`\) | 6 | \(`\omega_{23}`\) | 3 | |
| \(`\beta_7`\) | 5 | \(`\gamma_1`\) | 3 | |
| \(`\beta_8`\) | 8 | \(`\gamma_2`\) | 0 | |
| \(`\beta_9`\) | 9 | \(`\sigma`\) | 1.2 |
Here, the non-linear parameter \(`\theta`\) that can be included as a power on each \(`p_ip_j`\) component of each interaction variable (Connolly et al 2013) was set equal to one and thus does not appear in the equation above.
The 206 rows of proportions contained in the dataset
design_a (supplied in the package) were used to simulate
the sim3 dataset. Here is the first few rows from
design_a:
| community | richness | p1 | p2 | p3 | p4 | p5 | p6 | p7 | p8 | p9 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
Where community is an identifier for unique sets of
proportions and richness is the number of species in the
community.
The proportions in design_a were replicated over two
treatment levels, giving a total of 412 rows in the simulated dataset.
The sim3 data can be loaded and viewed in the usual
way.
data("sim3")
knitr::kable(head(sim3, 10))| community | richness | treatment | p1 | p2 | p3 | p4 | p5 | p6 | p7 | p8 | p9 | response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10.265 |
| 1 | 1 | B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 7.740 |
| 1 | 1 | A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 12.173 |
| 1 | 1 | B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 8.497 |
| 2 | 1 | A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 10.763 |
| 2 | 1 | B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 8.989 |
| 2 | 1 | A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 10.161 |
| 2 | 1 | B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 7.193 |
| 3 | 1 | A | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 10.171 |
| 3 | 1 | B | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 6.053 |
There are several graphical displays that will help to explore the data and it may also be useful to generate summary statistics.
hist(sim3$response, xlab = "Response", main = "")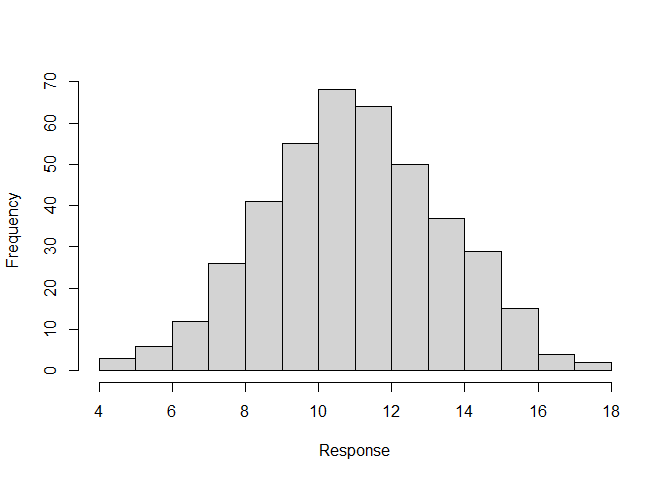
# Similar graphs can also be generated for the other species proportions.
plot(sim3$p1, sim3$response, xlab = "Proportion of species 1", ylab = "Response")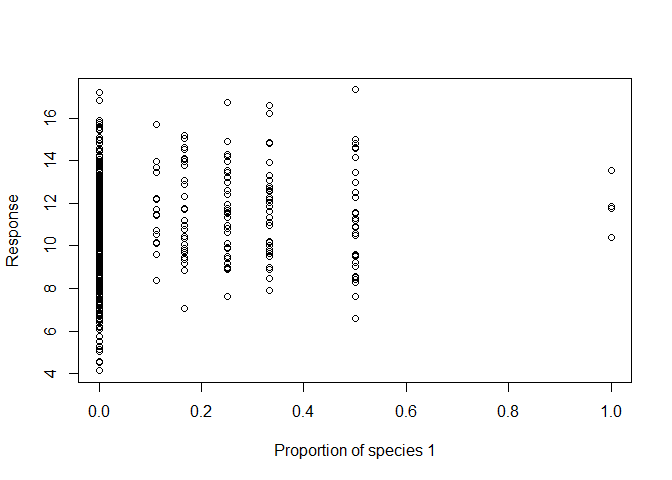
summary(sim3$response)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 4.134 9.327 10.961 10.994 12.604 17.323autoDIThe function autoDI in DImodels provides a
way to do an automated exploratory analysis to compare a range of DI
models. It works through a set of automated steps (Steps 1 to 4) and
will select the ‘best’ model from the range of models that have been
explored and test for lack of fit in that model. The selection process
is not exhaustive, but provides a useful starting point in analysis
using DI models.
auto1 <- autoDI(y = "response", prop = 4:12, treat = "treatment",
FG = c("FG1","FG1","FG1","FG1","FG1","FG2","FG2","FG3","FG3"), data = sim3,
selection = "Ftest")
#>
#> --------------------------------------------------------------------------------
#> Step 1: Investigating whether theta is equal to 1 or not for the AV model, including all available structures
#>
#> Theta estimate: 0.9714
#> Selection using F tests
#> Description
#> DI Model 1 Average interactions 'AV' DImodel with treatment
#> DI Model 2 Average interactions 'AV' DImodel with treatment, estimating theta
#>
#> DI_model treat estimate_theta Resid. Df Resid. SSq Resid. MSq
#> DI Model 1 AV 'treatment' FALSE 401 694.3095 1.7314
#> DI Model 2 AV 'treatment' TRUE 400 693.7321 1.7343
#> Df SSq F Pr(>F)
#> DI Model 1
#> DI Model 2 1 0.5775 0.333 0.5642
#>
#> The test concludes that theta is not significantly different from 1.
#>
#> --------------------------------------------------------------------------------
#> Step 2: Investigating the interactions
#> Since 'Ftest' was specified as selection criterion and functional groups were specified, dropping the ADD model as it is not nested within the FG model.
#> Selection using F tests
#> Description
#> DI Model 1 Structural 'STR' DImodel with treatment
#> DI Model 2 Species identity 'ID' DImodel with treatment
#> DI Model 3 Average interactions 'AV' DImodel with treatment
#> DI Model 4 Functional group effects 'FG' DImodel with treatment
#> DI Model 5 Separate pairwise interactions 'FULL' DImodel with treatment
#>
#> DI_model treat estimate_theta Resid. Df Resid. SSq Resid. MSq
#> DI Model 1 STR 'treatment' FALSE 410 1496.1645 3.6492
#> DI Model 2 ID 'treatment' FALSE 402 841.2740 2.0927
#> DI Model 3 AV 'treatment' FALSE 401 694.3095 1.7314
#> DI Model 4 FG 'treatment' FALSE 396 559.7110 1.4134
#> DI Model 5 FULL 'treatment' FALSE 366 522.9727 1.4289
#> Df SSq F Pr(>F)
#> DI Model 1
#> DI Model 2 8 654.8905 57.2903 <0.0001
#> DI Model 3 1 146.9645 102.8524 <0.0001
#> DI Model 4 5 134.5985 18.8396 <0.0001
#> DI Model 5 30 36.7383 0.857 0.686
#>
#> Selected model: Functional group effects 'FG' DImodel with treatment
#>
#> --------------------------------------------------------------------------------
#> Step 3: Investigating the treatment effect
#> Selection using F tests
#> Description
#> DI Model 1 Functional group effects 'FG' DImodel
#> DI Model 2 Functional group effects 'FG' DImodel with treatment
#>
#> DI_model treat estimate_theta Resid. Df Resid. SSq Resid. MSq
#> DI Model 1 FG none FALSE 397 1550.682 3.9060
#> DI Model 2 FG 'treatment' FALSE 396 559.711 1.4134
#> Df SSq F Pr(>F)
#> DI Model 1
#> DI Model 2 1 990.9711 701.12 <0.0001
#>
#> Selected model: Functional group effects 'FG' DImodel with treatment
#>
#> --------------------------------------------------------------------------------
#> Step 4: Comparing the final selected model with the reference (community) model
#> 'community' is a factor with 100 levels, one for each unique set of proportions.
#>
#> model Resid. Df Resid. SSq Resid. MSq Df SSq F Pr(>F)
#> DI Model 1 Selected 396 559.7110 1.4134
#> DI Model 2 Reference 311 445.9889 1.4340 85 113.7222 0.933 0.6423
#>
#> --------------------------------------------------------------------------------
#> autoDI is limited in terms of model selection. Exercise caution when choosing your final model.
#> --------------------------------------------------------------------------------The output of autoDI, works through the following
process:
AV) model and
uses profile likelihood to estimate the non-linear parameter \(`\theta`\) and tests whether or not it
differs from one. \(`\theta`\) was
estimated to be 0.96814 and was not significantly different from one
(\(`p = 0.4572`\)). Therefore,
subsequent steps assumed \(`\theta=1`\)
when fitting the DI models.Further details on each of these steps are available in the
autoDI help file. Run the following code to access the
documentation.
?autoDIAll parameter estimates from the selected model can be viewed using
summary.
summary(auto1)
#>
#> Call:
#> glm(formula = new_fmla, family = family, data = new_data)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> p1_ID 9.7497 0.3666 26.595 < 2e-16 ***
#> p2_ID 8.5380 0.3672 23.253 < 2e-16 ***
#> p3_ID 8.2329 0.3666 22.459 < 2e-16 ***
#> p4_ID 6.3644 0.3665 17.368 < 2e-16 ***
#> p5_ID 10.8468 0.3669 29.561 < 2e-16 ***
#> p6_ID 5.9621 0.4515 13.205 < 2e-16 ***
#> p7_ID 5.4252 0.4516 12.015 < 2e-16 ***
#> p8_ID 7.3204 0.4515 16.213 < 2e-16 ***
#> p9_ID 8.2154 0.4515 18.196 < 2e-16 ***
#> FG_bfg_FG1_FG2 3.4395 0.8635 3.983 8.09e-05 ***
#> FG_bfg_FG1_FG3 11.5915 0.8654 13.395 < 2e-16 ***
#> FG_bfg_FG2_FG3 2.8711 1.2627 2.274 0.02351 *
#> FG_wfg_FG1 2.8486 0.9131 3.120 0.00194 **
#> FG_wfg_FG2 0.6793 2.3553 0.288 0.77319
#> FG_wfg_FG3 2.4168 2.3286 1.038 0.29997
#> treatmentA 3.1018 0.1171 26.479 < 2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> (Dispersion parameter for gaussian family taken to be 1.413412)
#>
#> Null deviance: 52280.33 on 412 degrees of freedom
#> Residual deviance: 559.71 on 396 degrees of freedom
#> AIC: 1329.4
#>
#> Number of Fisher Scoring iterations: 2If the final model selected by autoDI includes a value of theta other
than 1, then a 95% confidence interval for \(`\theta`\) can be generated using the
theta_CI function:
theta_CI(auto1, conf = .95)Here, this code would not run, since the final model selected by
autoDI does not include theta estimated.
DI functionFor some users, the selection process in autoDI will be
sufficient, however, most users will fit additional models using
DI. For example, while the treatment is included in
autoDI as an additive factor, interactions between
treatment and other model terms are not considered. Here, we will first
fit the model selected by autoDI using DI and
then illustrate the capabilities of DI to fit specialised
models.
autoDI using DIm1 <- DI(y = "response", prop = 4:12,
FG = c("FG1","FG1","FG1","FG1","FG1","FG2","FG2","FG3","FG3"), treat = "treatment",
DImodel = "FG", data = sim3)
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Functional group effects 'FG' DImodel
summary(m1)
#>
#> Call:
#> glm(formula = new_fmla, family = family, data = new_data)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> p1_ID 9.7497 0.3666 26.595 < 2e-16 ***
#> p2_ID 8.5380 0.3672 23.253 < 2e-16 ***
#> p3_ID 8.2329 0.3666 22.459 < 2e-16 ***
#> p4_ID 6.3644 0.3665 17.368 < 2e-16 ***
#> p5_ID 10.8468 0.3669 29.561 < 2e-16 ***
#> p6_ID 5.9621 0.4515 13.205 < 2e-16 ***
#> p7_ID 5.4252 0.4516 12.015 < 2e-16 ***
#> p8_ID 7.3204 0.4515 16.213 < 2e-16 ***
#> p9_ID 8.2154 0.4515 18.196 < 2e-16 ***
#> FG_bfg_FG1_FG2 3.4395 0.8635 3.983 8.09e-05 ***
#> FG_bfg_FG1_FG3 11.5915 0.8654 13.395 < 2e-16 ***
#> FG_bfg_FG2_FG3 2.8711 1.2627 2.274 0.02351 *
#> FG_wfg_FG1 2.8486 0.9131 3.120 0.00194 **
#> FG_wfg_FG2 0.6793 2.3553 0.288 0.77319
#> FG_wfg_FG3 2.4168 2.3286 1.038 0.29997
#> treatmentA 3.1018 0.1171 26.479 < 2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> (Dispersion parameter for gaussian family taken to be 1.413412)
#>
#> Null deviance: 52280.33 on 412 degrees of freedom
#> Residual deviance: 559.71 on 396 degrees of freedom
#> AIC: 1329.4
#>
#> Number of Fisher Scoring iterations: 2autoDI estimating theta using
update_DIm1_theta <- update_DI(object = m1, estimate_theta = TRUE)
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Functional group effects 'FG' DImodel
#> Theta estimate: 0.9681
coef(m1_theta)
#> p1_ID p2_ID p3_ID p4_ID p5_ID
#> 9.8128865 8.6069092 8.2968619 6.4287580 10.9110563
#> p6_ID p7_ID p8_ID p9_ID FG_bfg_FG1_FG2
#> 6.0189395 5.4846833 7.4038925 8.2992262 2.9840924
#> FG_bfg_FG1_FG3 FG_bfg_FG2_FG3 FG_wfg_FG1 FG_wfg_FG2 FG_wfg_FG3
#> 10.6019235 2.3514998 2.3737831 0.3789464 1.8470612
#> treatmentA theta
#> 3.1017864 0.9681005The species identity effects in a DI model can be grouped by
specifying groups for each species using the ID argument.
The ID argument functions similar to the FG
argument and accepts a character list of same length as number of
species in the model. The identity effects of species belonging in the
same group will be grouped together.
Grouping all identity effects into a single term
m1_group <- update_DI(object = m1_theta,
ID = c("ID1", "ID1", "ID1", "ID1", "ID1",
"ID1", "ID1", "ID1", "ID1"))
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Functional group effects 'FG' DImodel
#> Theta estimate: 0.9919
coef(m1_group)
#> ID1 FG_bfg_FG1_FG2 FG_bfg_FG1_FG3 FG_bfg_FG2_FG3 FG_wfg_FG1
#> 7.8667702 1.1475018 12.9438529 -1.2235215 5.6141823
#> FG_wfg_FG2 FG_wfg_FG3 treatmentA theta
#> -5.5214662 1.0205019 3.1017864 0.9919097Grouping identity effects of specific species
m1_group2 <- update_DI(object = m1_theta,
ID = c("ID1", "ID1", "ID1",
"ID2", "ID2", "ID2",
"ID3", "ID3", "ID3"))
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Functional group effects 'FG' DImodel
#> Theta estimate: 0.989
coef(m1_group2)
#> ID1 ID2 ID3 FG_bfg_FG1_FG2 FG_bfg_FG1_FG3
#> 8.5288216 7.9537767 7.1357104 0.9665077 13.3434768
#> FG_bfg_FG2_FG3 FG_wfg_FG1 FG_wfg_FG2 FG_wfg_FG3 treatmentA
#> 0.4940952 4.1543637 -4.4683501 3.4674196 3.1017864
#> theta
#> 0.9889999Note: Grouping ID effects will not have an effect on the calculation of the interaction effects, they would still be calculated by using all species.
Read the documentation of DI and autoDI for
more information and examples using the ID parameter.
?DI
?autoDIDI functionThere are two ways to fit customised models using DI;
the first is by using the option DImodel = in the
DI function and adding the argument
extra_formula = to it, and the second is to use the
custom_formula argument in the DI function. If
species interaction variables (e.g., the FG interactions or the average
pairwise interaction) are included in either extra_formula
or custom_formula, they must first be created and included
in the dataset. The function DI_data can be used to compute
several types of species interaction variables.
extra_formulam2 <- DI(y = "response", prop = 4:12,
FG = c("FG1","FG1","FG1","FG1","FG1","FG2","FG2","FG3","FG3"), treat = "treatment",
DImodel = "FG", extra_formula = ~ (p1 + p2 + p3 + p4):treatment,
data = sim3)
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Functional group effects 'FG' DImodel
summary(m2)
#>
#> Call:
#> glm(formula = new_fmla, family = family, data = new_data)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> p1_ID 10.018491 0.466552 21.473 < 2e-16 ***
#> p2_ID 8.494038 0.467009 18.188 < 2e-16 ***
#> p3_ID 7.970716 0.466536 17.085 < 2e-16 ***
#> p4_ID 6.624476 0.466443 14.202 < 2e-16 ***
#> p5_ID 10.802270 0.378776 28.519 < 2e-16 ***
#> p6_ID 5.917565 0.461482 12.823 < 2e-16 ***
#> p7_ID 5.380703 0.461535 11.658 < 2e-16 ***
#> p8_ID 7.275881 0.461506 15.766 < 2e-16 ***
#> p9_ID 8.170907 0.461471 17.706 < 2e-16 ***
#> FG_bfg_FG1_FG2 3.439508 0.865279 3.975 8.38e-05 ***
#> FG_bfg_FG1_FG3 11.591458 0.867140 13.367 < 2e-16 ***
#> FG_bfg_FG2_FG3 2.871063 1.265295 2.269 0.02381 *
#> FG_wfg_FG1 2.848612 0.915008 3.113 0.00199 **
#> FG_wfg_FG2 0.679285 2.360195 0.288 0.77365
#> FG_wfg_FG3 2.416774 2.333420 1.036 0.30097
#> treatmentA 3.190868 0.216493 14.739 < 2e-16 ***
#> `treatmentA:p1` -0.626667 0.668369 -0.938 0.34902
#> `treatmentA:p2` -0.001213 0.668369 -0.002 0.99855
#> `treatmentA:p3` 0.435322 0.668369 0.651 0.51522
#> `treatmentA:p4` -0.609180 0.668369 -0.911 0.36262
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> (Dispersion parameter for gaussian family taken to be 1.419257)
#>
#> Null deviance: 52280.33 on 412 degrees of freedom
#> Residual deviance: 556.35 on 392 degrees of freedom
#> AIC: 1335
#>
#> Number of Fisher Scoring iterations: 2extra_formulaFirst, we create the FG pairwise interactions, using the
DI_data function with the what argument set to
"FG".
FG_matrix <- DI_data(prop = 4:12, FG = c("FG1","FG1","FG1","FG1","FG1","FG2","FG2","FG3","FG3"),
data = sim3, what = "FG")
sim3a <- data.frame(sim3, FG_matrix)Then we fit the model using extra_formula.
m3 <- DI(y = "response", prop = 4:12,
FG = c("FG1","FG1","FG1","FG1","FG1","FG2","FG2","FG3","FG3"),
treat = "treatment", DImodel = "FG",
extra_formula = ~ (bfg_FG1_FG2 + bfg_FG1_FG3 + bfg_FG2_FG3 +
wfg_FG1 + wfg_FG2 + wfg_FG3) : treatment, data = sim3a)
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Functional group effects 'FG' DImodel
summary(m3)
#>
#> Call:
#> glm(formula = new_fmla, family = family, data = new_data)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> p1_ID 9.68668 0.40000 24.217 < 2e-16 ***
#> p2_ID 8.47495 0.40053 21.159 < 2e-16 ***
#> p3_ID 8.16990 0.39998 20.426 < 2e-16 ***
#> p4_ID 6.30140 0.39987 15.759 < 2e-16 ***
#> p5_ID 10.78379 0.40031 26.938 < 2e-16 ***
#> p6_ID 5.89908 0.47958 12.301 < 2e-16 ***
#> p7_ID 5.36222 0.47963 11.180 < 2e-16 ***
#> p8_ID 7.25740 0.47960 15.132 < 2e-16 ***
#> p9_ID 8.15243 0.47957 17.000 < 2e-16 ***
#> FG_bfg_FG1_FG2 4.00191 1.12383 3.561 0.000415 ***
#> FG_bfg_FG1_FG3 11.77389 1.12973 10.422 < 2e-16 ***
#> FG_bfg_FG2_FG3 3.83681 1.64287 2.335 0.020027 *
#> FG_wfg_FG1 2.81860 1.16226 2.425 0.015757 *
#> FG_wfg_FG2 -1.58378 3.11717 -0.508 0.611682
#> FG_wfg_FG3 1.32358 3.07561 0.430 0.667181
#> treatmentA 3.22783 0.33480 9.641 < 2e-16 ***
#> `treatmentA:bfg_FG1_FG2` -1.12480 1.43053 -0.786 0.432178
#> `treatmentA:bfg_FG1_FG3` -0.36487 1.44450 -0.253 0.800717
#> `treatmentA:bfg_FG2_FG3` -1.93150 2.09024 -0.924 0.356029
#> `treatmentA:wfg_FG1` 0.06003 1.42911 0.042 0.966517
#> `treatmentA:wfg_FG2` 4.52613 4.06260 1.114 0.265924
#> `treatmentA:wfg_FG3` 2.18638 3.99748 0.547 0.584733
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> (Dispersion parameter for gaussian family taken to be 1.42436)
#>
#> Null deviance: 52280.3 on 412 degrees of freedom
#> Residual deviance: 555.5 on 390 degrees of freedom
#> AIC: 1338.3
#>
#> Number of Fisher Scoring iterations: 2custom_formulaFirst, we create a dummy variable for level A of the treatment (this
is required for the glm engine that is used within
DI and because there is no intercept in the model).
sim3a$treatmentA <- as.numeric(sim3a$treatment == "A")Then we fit the model using custom_formula.
m3 <- DI(y = "response",
custom_formula = response ~ 0 + p1 + p2 + p3 + p4 + p5 + p6 + p7 + p8 + p9 +
treatmentA + bfg_FG1_FG2 + bfg_FG1_FG3 + bfg_FG2_FG3, data = sim3a)
#> Fitted model: Custom DI model
summary(m3)
#>
#> Call:
#> glm(formula = custom_formula, family = family, data = data)
#>
#> Coefficients:
#> Estimate Std. Error t value Pr(>|t|)
#> p1 10.3417 0.3138 32.957 < 2e-16 ***
#> p2 9.1766 0.3103 29.573 < 2e-16 ***
#> p3 8.8268 0.3134 28.164 < 2e-16 ***
#> p4 6.9742 0.3122 22.341 < 2e-16 ***
#> p5 11.4422 0.3141 36.426 < 2e-16 ***
#> p6 5.9177 0.3994 14.815 < 2e-16 ***
#> p7 5.3967 0.3999 13.496 < 2e-16 ***
#> p8 7.4468 0.3983 18.695 < 2e-16 ***
#> p9 8.3449 0.3984 20.945 < 2e-16 ***
#> treatmentA 3.1018 0.1184 26.198 < 2e-16 ***
#> bfg_FG1_FG2 2.9359 0.8042 3.651 0.000296 ***
#> bfg_FG1_FG3 10.8896 0.8343 13.053 < 2e-16 ***
#> bfg_FG2_FG3 2.9410 1.2233 2.404 0.016667 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#>
#> (Dispersion parameter for gaussian family taken to be 1.443887)
#>
#> Null deviance: 52280.33 on 412 degrees of freedom
#> Residual deviance: 576.11 on 399 degrees of freedom
#> AIC: 1335.3
#>
#> Number of Fisher Scoring iterations: 2We can make predictions from a DI model just like any other
regression model using the predict function. The user does
not need to worry about adding any interaction terms or adjusting any
columns if theta is not equal to 1. Only the species proportions along
with any additional experimental structures is needed and all other
terms in the model will be calculated for the user.
# Fit model
m3 <- DI(y = "response", prop = 4:12,
treat = "treatment", DImodel = "AV",
extra_formula = ~ (AV) : treatment, data = sim3a)
#> Warning in DI_data_prepare(y = y, block = block, density = density, prop = prop, : One or more rows have species proportions that sum to approximately 1, but not exactly 1. This is typically a rounding issue, and has been corrected internally prior to analysis.
#> Fitted model: Average interactions 'AV' DImodel
predict_data <- sim3[c(1, 79, 352), 3:12]
# Only species proportions and treatment is needed
print(predict_data)
#> treatment p1 p2 p3 p4 p5 p6 p7 p8
#> 1 A 0 0 0.0000000 0 0.0000000 0.0000000 0.0000000 0.0000000
#> 79 A 0 0 0.0000000 0 0.5000000 0.0000000 0.0000000 0.5000000
#> 352 B 0 0 0.1666667 0 0.1666667 0.1666667 0.1666667 0.1666667
#> p9
#> 1 1.0000000
#> 79 0.0000000
#> 352 0.1666667
# Make prediction
predict(m3, newdata = predict_data)
#> 1 79 352
#> 12.83789 14.27503 10.00291# The interval and level parameters can be used to calculate the
# uncertainty around the predictions
# Get confidence interval around prediction
predict(m3, newdata = predict_data, interval = "confidence")
#> fit lwr upr
#> 1 12.83789 12.028716 13.64707
#> 79 14.27503 13.817612 14.73246
#> 352 10.00291 9.694552 10.31126
# Get prediction interval around prediction
predict(m3, newdata = predict_data, interval = "prediction")
#> fit lwr upr
#> 1 12.83789 10.124779 15.55100
#> 79 14.27503 11.645310 16.90476
#> 352 10.00291 7.394976 12.61083
# The function returns a 95% interval by default,
# this can be changed using the level argument
predict(m3, newdata = predict_data,
interval = "prediction", level = 0.9)
#> fit lwr upr
#> 1 12.83789 10.562595 15.11319
#> 79 14.27503 12.069670 16.48040
#> 352 10.00291 7.815819 12.18999The contrasts_DI function can be used to compare and
formally test for a difference in performance of communities within the
same as well as across different experimental structures.
The contrast_vars parameter can be used to quickly
calculate contrasts without having to calculate interaction terms
Comparing the performance of the monocultures of different species at
treatment A
contr <- data.frame(p1 = c(1, 0),
p2 = c(-1, 0),
p7 = c(0, 1),
p9 = c(0, -1))
rownames(contr) <- c("p1_vs_p2", "p7_vs_p9")
the_C <- contrasts_DI(m3, contrast_vars = contr)
#> Warning in add_extra_vars(object, newdata = the_C): treatment not supplied in
#> newdata. Calculating for 'treatment' = A
#> Generated contrast matrix:
#> p1_ID p2_ID p3_ID p4_ID p5_ID p6_ID p7_ID p8_ID p9_ID AV treatmentA
#> p1_vs_p2 1 -1 0 0 0 0 0 0 0 0 1
#> p7_vs_p9 0 0 0 0 0 0 1 0 -1 0 1
#> AV:treatmentB
#> p1_vs_p2 0
#> p7_vs_p9 0
summary(the_C)
#>
#> Simultaneous Tests for General Linear Hypotheses
#>
#> Fit: glm(formula = new_fmla, family = family, data = new_data)
#>
#> Linear Hypotheses:
#> Estimate Std. Error z value Pr(>|z|)
#> p1_vs_p2 == 0 4.7010 0.6034 7.791 < 1e-10 ***
#> p7_vs_p9 == 0 -2.2931 0.6034 -3.800 0.000288 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#> (Adjusted p values reported -- single-step method)Comparing across the two treatment levels
contr <- data.frame("treatmentA" = 1)
rownames(contr) <- "p1_TreatmentAvsB"
the_C <- contrasts_DI(m3, contrast_vars = contr)
#> Warning in add_extra_vars(object, newdata = the_C): treatment not supplied in
#> newdata. Calculating for 'treatment' = A
#> Generated contrast matrix:
#> p1_ID p2_ID p3_ID p4_ID p5_ID p6_ID p7_ID p8_ID p9_ID AV
#> p1_TreatmentAvsB 0 0 0 0 0 0 0 0 0 0
#> treatmentA AV:treatmentB
#> p1_TreatmentAvsB 1 0
summary(the_C)
#>
#> Simultaneous Tests for General Linear Hypotheses
#>
#> Fit: glm(formula = new_fmla, family = family, data = new_data)
#>
#> Linear Hypotheses:
#> Estimate Std. Error z value Pr(>|z|)
#> p1_TreatmentAvsB == 0 3.2278 0.3695 8.735 <2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#> (Adjusted p values reported -- single-step method)Comparing between two species mixtures
# Suppose these are species communities we wish to compare
mixA <- c(0.25, 0, 0.25, 0, 0.25, 0, 0.25, 0, 0)
mixB <- c(0, 0.3333, 0, 0.3333, 0, 0.3333, 0, 0, 0)
# The contrast can be created by subtracting the species proportions
contr <- matrix(mixA - mixB, nrow = 1)
colnames(contr) <- paste0("p", 1:9)
print(contr)
#> p1 p2 p3 p4 p5 p6 p7 p8 p9
#> [1,] 0.25 -0.3333 0.25 -0.3333 0.25 -0.3333 0.25 0 0
# The values for the interaction terms will be calculated
# automatically
the_C <- contrasts_DI(m3, contrast_vars = contr)
#> Warning in add_extra_vars(object, newdata = the_C): treatment not supplied in
#> newdata. Calculating for 'treatment' = A
#> Generated contrast matrix:
#> p1_ID p2_ID p3_ID p4_ID p5_ID p6_ID p7_ID p8_ID p9_ID AV
#> `Test 1` 0.25 -0.3333 0.25 -0.3333 0.25 -0.3333 0.25 0 0 0.04173333
#> treatmentA AV:treatmentB
#> `Test 1` 1 0
summary(the_C)
#>
#> Simultaneous Tests for General Linear Hypotheses
#>
#> Fit: glm(formula = new_fmla, family = family, data = new_data)
#>
#> Linear Hypotheses:
#> Estimate Std. Error z value Pr(>|z|)
#> `Test 1` == 0 5.2658 0.4327 12.17 <2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#> (Adjusted p values reported -- single-step method)Additionally, the contrast_matrix function provides
flexibility to manually create the contrast matrix with all the
identity, interaction and any additional terms in the model. This output
can then be passed onto the contrast parameter in
contrasts_DI to test for contrasts
# p1, p2, p5 and p7 equi-proportional mixture at treatment A
mixA <- contrast_matrix(object = m3,
contrast_vars = data.frame("p1" = 0.25,
"p2" = 0.25, "p5" = 0.25,
"p7" = 0.25,
"treatmentA" = 1))
#> Warning in add_extra_vars(object, newdata = the_C): treatment not supplied in
#> newdata. Calculating for 'treatment' = A
# p2, p4, and p6 equi-proportional mixture at treatment A
mixB <- contrast_matrix(object = m3,
contrast_vars = data.frame("p2" = 1/3,
"p4" = 1/3, "p6" = 1/3,
"treatmentA" = 1))
#> Warning in add_extra_vars(object, newdata = the_C): treatment not supplied in
#> newdata. Calculating for 'treatment' = A
# Subtracting these two values would give us the contrast for
# comparing these mixtures
my_contrast <- mixA - mixB
rownames(my_contrast) <- "4_sp_mix vs 3_sp_mix"
# This contrast can be passed to the `contrast` parameter in `contrasts_DI`
the_C <- contrasts_DI(m3, contrast = my_contrast)
#> Generated contrast matrix:
#> p1_ID p2_ID p3_ID p4_ID p5_ID p6_ID p7_ID
#> 4_sp_mix vs 3_sp_mix 0.25 -0.08333333 0 -0.3333333 0.25 -0.3333333 0.25
#> p8_ID p9_ID AV treatmentA AV:treatmentB
#> 4_sp_mix vs 3_sp_mix 0 0 0.04166667 0 0
summary(the_C)
#>
#> Simultaneous Tests for General Linear Hypotheses
#>
#> Fit: glm(formula = new_fmla, family = family, data = new_data)
#>
#> Linear Hypotheses:
#> Estimate Std. Error z value Pr(>|z|)
#> 4_sp_mix vs 3_sp_mix == 0 2.0507 0.2204 9.303 <2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#> (Adjusted p values reported -- single-step method)The compare_communities function can be used to perform
pairwise comparisons of communities and assign CLD to each community.
The function accepts a fitted DI model object and a
data.frame containing the proportions of communities to be
compared and optionally, values for any additional treatment
variables.
comms_to_compare <- sim3[c(36, 79, 352), 3:12]
compare_communities(m3, data = comms_to_compare)
#> $Contrasts
#>
#> Simultaneous Tests for General Linear Hypotheses
#>
#> Fit: glm(formula = new_fmla, family = family, data = new_data)
#>
#> Linear Hypotheses:
#> Estimate Std. Error z value Pr(>|z|)
#> 36-79 == 0 -4.6036 0.4988 -9.229 <1e-05 ***
#> 36-352 == 0 -0.3314 0.5010 -0.661 0.778
#> 79-352 == 0 4.2721 0.2571 16.617 <1e-05 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#> (Adjusted p values reported -- single-step method)
#>
#>
#> $CLD
#> 36 79 352
#> "a" "b" "a"The function returns a list containing the pairwise contrasts and CLD symbol for each community.
If all possible pairwise comparisons are not desired, the
ref argument can be used to compare all communities against
a specific reference. Additionally, the adjust argument can
be used to choose a particular adjustment method for calculating the
p-values.
# ref accepts either a row-number of rowname of the community to be used as reference
# adjust should be one of the following
# c("single-step", "Shaffer", "Westfall", "free", "holm",
# "hochberg", "hommel", "bonferroni", "BH", "BY", "fdr", "none")
compare_communities(m3, data = comms_to_compare,
ref = 3, adjust = "bonferroni")
#> $Contrasts
#>
#> Simultaneous Tests for General Linear Hypotheses
#>
#> Fit: glm(formula = new_fmla, family = family, data = new_data)
#>
#> Linear Hypotheses:
#> Estimate Std. Error z value Pr(>|z|)
#> 36-352 == 0 -0.3314 0.5010 -0.661 1
#> 79-352 == 0 4.2721 0.2571 16.617 <2e-16 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
#> (Adjusted p values reported -- bonferroni method)
#>
#>
#> $CLD
#> 352 (ref) 36 79
#> "a" "a" "b"Connolly J, T Bell, T Bolger, C Brophy, T Carnus, JA Finn, L Kirwan, F Isbell, J Levine, A Lüscher, V Picasso, C Roscher, MT Sebastia, M Suter and A Weigelt (2013) An improved model to predict the effects of changing biodiversity levels on ecosystem function. Journal of Ecology, 101, 344-355.
Kirwan L, J Connolly, JA Finn, C Brophy, A Lüscher, D Nyfeler and MT Sebastia (2009) Diversity-interaction modelling - estimating contributions of species identities and interactions to ecosystem function. Ecology, 90, 2032-2038.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.