The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
![]()
As of version 2.0.0 the MIC package has being refocused to provide
MIC-specific analysis and validation utilities. Genomics-related
functions (PATRIC download helpers, genome -> \(k\)-mer conversion, \(k\)-mer utilities, and similar) have been
ported to a new package, faLearn.
To continue using these functions, simply install and load the
faLearn package alongside MIC:
# install.packages("remotes")
remotes::install_github("agerada/faLearn")
library(faLearn)MIC is an R package for the analysis of minimum
inhibitory concentration (MIC) data. The package was designed to be
compatible with the AMR, in particular most
of the functions in MIC are designed to accept and return
AMR objects, such as mic and sir.
The primary functions in MIC are designed towards
validation studies of minimum inhibitory concentrations, however it also
can (optionally) be used to support the construction of machine learning
models that predict MIC values from genomic data.
Rcpp.XGBoost-compatible
libsvm format.install.packages("MIC")# install.packages("remotes")
remotes::install_github("agerada/MIC")Load the MIC package – it is highly recommended that
AMR is also loaded. Where possible, MIC
functions maintain compatibility with AMR objects, in
particular the mic and sir classes.
library(MIC)
#>
#> Attaching package: 'MIC'
#> The following object is masked from 'package:base':
#>
#> table
library(AMR)To compare two mic vectors (e.g., one from a gold
standard and one from a prediction or investigational assay), the
compare_mic function can be used. An example dataset of MIC
values is provided with the package, which will be used here.
data("example_mics")
head(example_mics)
#> gs test mo ab
#> 1 0.002 0.002 B_ESCHR_COLI GEN
#> 2 0.004 0.002 B_ESCHR_COLI GEN
#> 3 8 16 B_ESCHR_COLI GEN
#> 4 0.008 0.016 B_ESCHR_COLI GEN
#> 5 64 64 B_ESCHR_COLI GEN
#> 6 0.06 0.06 B_ESCHR_COLI GENThe dataset contains MIC values (in mic format) for a
“test” assay, and a “gold standard” (gs) assay. We will use
compare_mic to compare the MICs and validate the “test”
assay:
val <- compare_mic(gold_standard = example_mics$gs, test = example_mics$test)
val
#> MIC validation object with 300 observations
#> Agreement type: essentialCalling summary provides the essential agreement (EA)
rates and assay bias:
summary(val)
#> MIC validation summary
#> Essential agreement: 267 (89%)
#> Bias: -7If organisms and antimicrobials are provided,
compare_mic will also calculate and return the categorical
agreement (CA) rates, in the form of minor, major, and very major
errors:
val <- compare_mic(gold_standard = example_mics$gs, test = example_mics$test,
mo = example_mics$mo, ab = example_mics$ab)
val
#> MIC validation object with 300 observations
#> Agreement type: essential and categorical
#> Antibiotics: GEN, MEM, AMX
#> Organisms: B_ESCHR_COLIThis time, calling summary will provide a breakdown of
the categorical agreement rates in addition to the EA rates:
summary(val)
#> MIC validation summary
#> Antibiotic: AMX, GEN, MEM
#> Organism: B_ESCHR_COLI
#> Essential agreement: 267 (89%)
#> Resistant: 113 (37.67%)
#> Minor errors: 0 (0%)
#> Major errors: 6 (2%)
#> Very major errors: 8 (2.67%)
#> Mean bias: -7
#> N: 300
#> *Use as.data.frame() to see full summary*Using as.data.frame allows us to continue working with
the summarised results:
head(as.data.frame(val))
#> gold_standard test essential_agreement ab mo gold_standard_sir
#> 1 0.002 0.002 TRUE GEN B_ESCHR_COLI S
#> 2 0.004 0.002 TRUE GEN B_ESCHR_COLI S
#> 3 8 16 TRUE GEN B_ESCHR_COLI R
#> 4 0.008 0.016 TRUE GEN B_ESCHR_COLI S
#> 5 64 64 TRUE GEN B_ESCHR_COLI R
#> 6 0.06 0.06 TRUE GEN B_ESCHR_COLI S
#> test_sir error
#> 1 S <NA>
#> 2 S <NA>
#> 3 R <NA>
#> 4 S <NA>
#> 5 R <NA>
#> 6 S <NA>The results of an mic_validation can be plotted in a
confusion matrix (failed essential agreements are in red):
plot(val)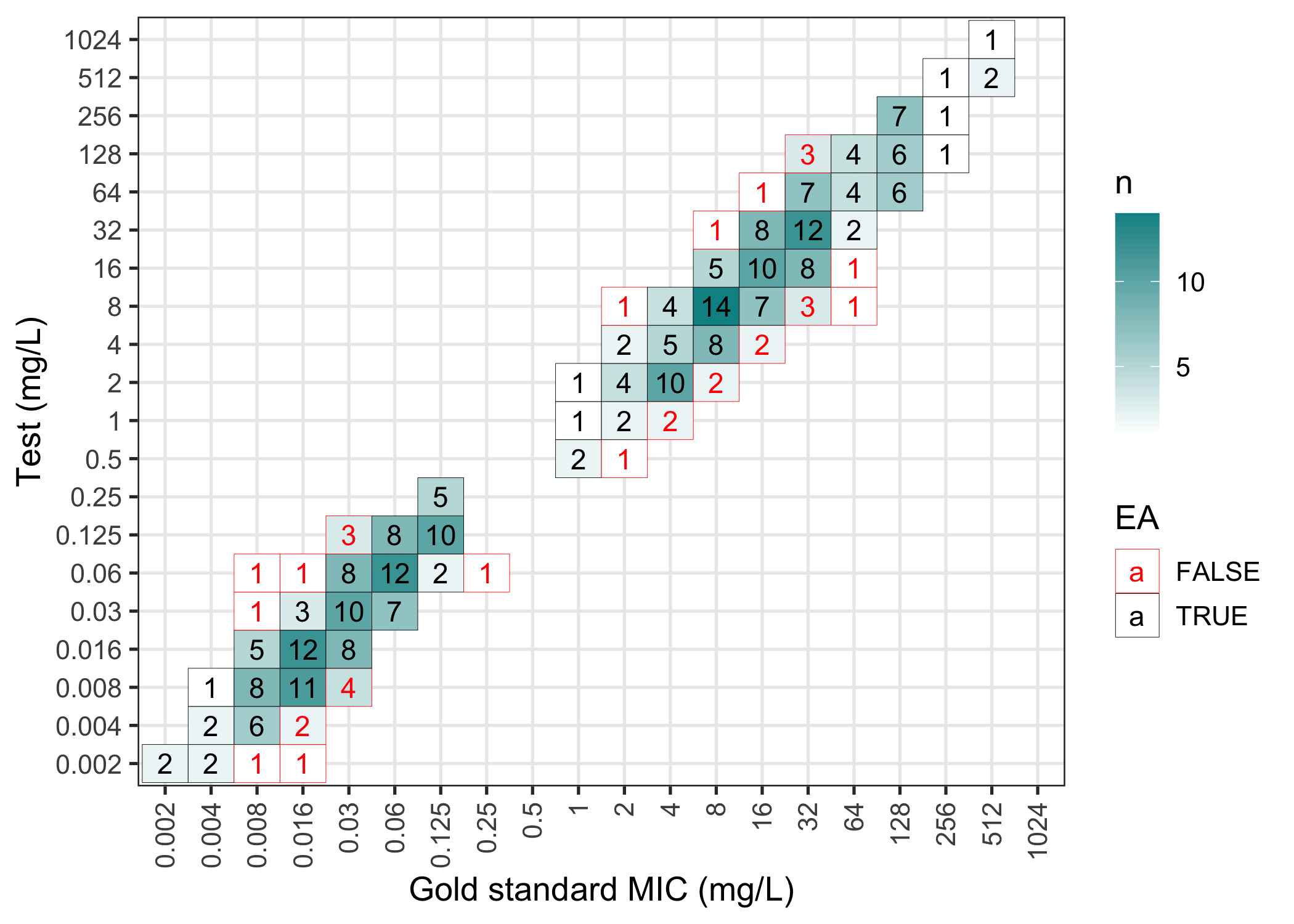
The plot can also be faceted by antimicrobial:
plot(val, facet_wrap_ncol = 1)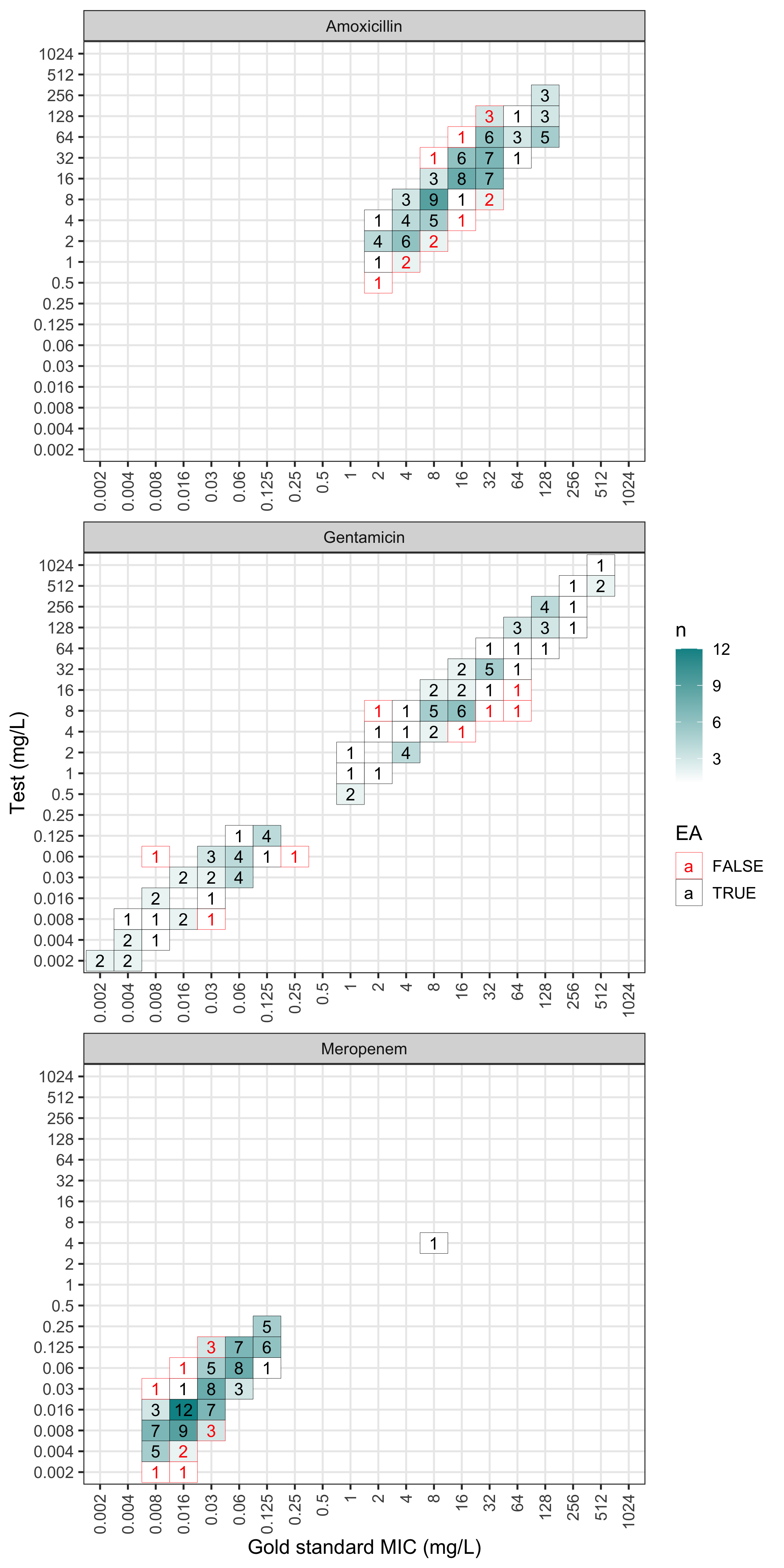
The table function can be used to generate a table of
the results:
# generate table for MEM
mem_dat <- subset(example_mics, ab == "MEM")
mem_val <- compare_mic(gold_standard = mem_dat$gs, test = mem_dat$test)
table(mem_val)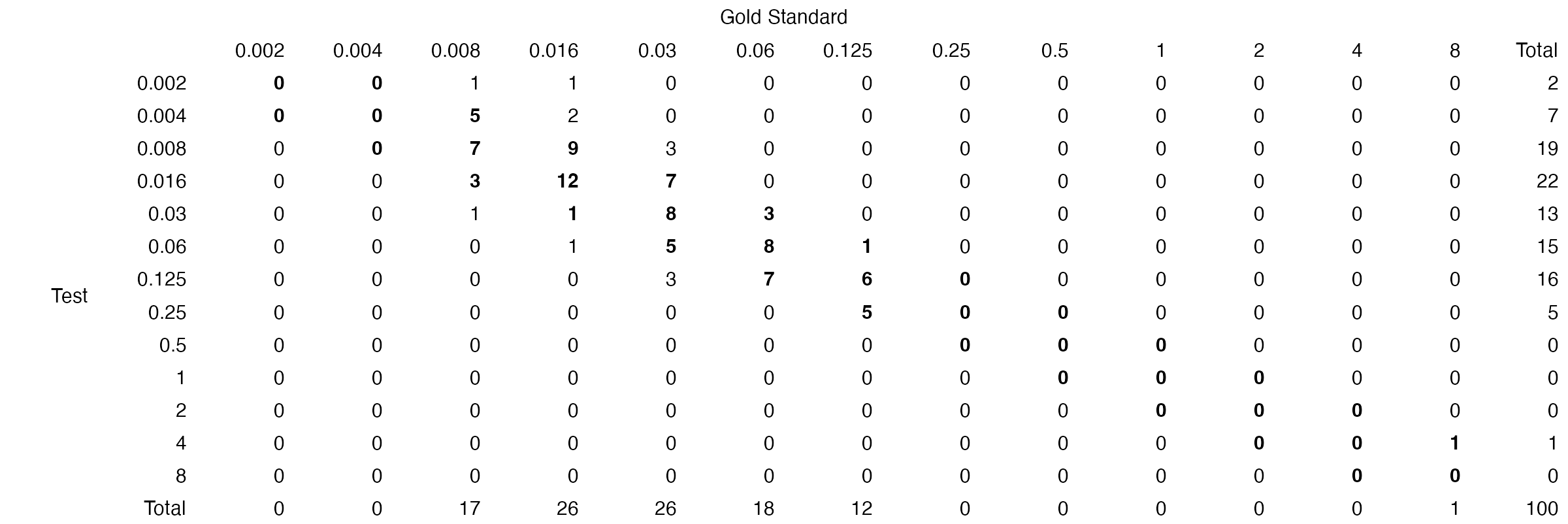
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.