


The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
Metagenome taxonomy assignment comparison toolkit. The toolkit is being developed for EDGE platform and reflects its backend specificity. The routines, however, can be used as a stand-alone library for multi-project comparative visualization of taxonomy assignments obtained for metagenomic samples processed with GOTTCHA/GOTTCHA2, BWA, KRAKEN, METAPHLAN, DIAMOND, or PANGIA. The heatmaps can be also visualized with this D3.js-based code which allows to see the exact abundance values in each cell.
install.packages("MetaComp")to use the library, simply load it into R environment:
library(MetaComp)install.packages("devtools")
library(devtools)
install_github(repo = 'seninp-bioinfo/MetaComp')the_gottcha2_assignment <- load_edge_assignment(data_file_g2, type = 'gottcha2')
the_kraken_assignment <- load_edge_assignment(data_file_k, type = 'kraken')
the_pangia_assignment <- load_edge_assignment(data_file_p, type = 'pangia')The package functions load_xxx_assignments (where
xxx stands for gottcha, kraken, or metaphlan) are designed
to read a tool-specific assignment files. The configuration file for
these functions must be tab-delimeted two columns file where the first
column is the project id (used as the project’s name in plotting), and
the second column is an actual assignment file path:
the_assignments_list_g2 <- load_edge_assignments(config_file_g2, type = 'gottcha2')
the_assignments_list_k <- load_edge_assignments(config_file_k, type = 'kraken')
the_assignments_list_p <- load_edge_assignments(config_file_pangia, type = 'pangia')The merge_edge_assignments function is capable to merge
a named list of GOTTCHA, Kraken, or MetaPhlAn assignments into a single
table using LEVEL and TAXA columns as ids.
The function plot_edge_assignment accepts a single
assignment table and outputs a ggplot object or produces a PDF plot
using ggplot2’s geom_tile.
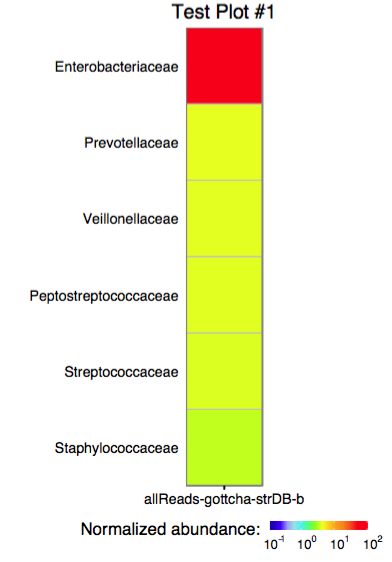
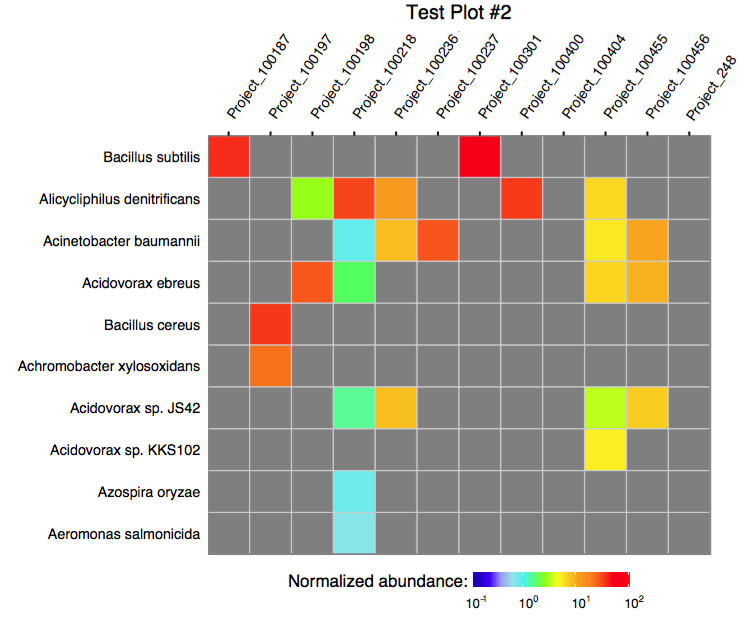
The function plot_merged_assignment accepts a single
merged assignment table as an input and outputs a ggplot object or
produces a PDF plot using ggplot2’s geom_tile.
The following script can be used to run the merge procedure in a batch mode:
# load library
require(MetaComp)
#
# configure runtime
options(echo = TRUE)
args <- commandArgs(trailingOnly = TRUE)
#
# print provided args
print(paste("provided args: ", args))
#
# acquire values
srcFile <- args[1]
destFile <- args[2]
taxonomyLevelArg <- args[3]
plotTitleArg <- args[4]
plotFileArg <- args[5]
#
# extended functionality was added in the release #3, and we don't want to break the legacy systems
#
if (length(args) > 5) {
rowLimitArg <- args[6]
sortingOrderArg <- args[7]
} else {
rowLimitArg <- 60
sortingOrderArg <- "abundance"
}
#
# read the data and produce the merged table
merged <- merge_edge_assignments(load_edge_assignments(srcFile, type = "gottcha2"))
#
# write the merge table as a TAB-delimeted file
write.table(merged, file = destFile, col.names = T, row.names = F, quote = T, sep = "\t")
#
# produce a PDF of the merged assignment
plot_merged_assignment(assignment = merged, taxonomy_level = taxonomyLevelArg,
sorting_order = sortingOrderArg, row_limit = base::strtoi(rowLimitArg),
plot_title = plotTitleArg, filename = plotFileArg)To execute the scrip, use Rscript as shown below:
$> Rscript merge_and_plot_gottcha_assignments.R assignments_table_gottcha.txt merged_assignments.txt \
family "Merge test plot" merge_test 20 alphabeticalthis command line arguments are (some of these are clickable – so you
can see examples): * Rscript - a way to execute the R
script * merge_and_plot_gottcha_assignments.R-
the above script filename * assignments_table_gottcha.txt
- the tab delimeted table of assignments (two columns:
project_id TAB assignment_path) * merged_assignments_gottcha.txt
- the tab-delimeted output file name * family - a LEVEL at
which the plot should be produced * "Merge test plot"- the
output plot’s title * merge_test - the output plot filename
mask, ".pdf"
and ".svg"
files will be produced… * 20 the max number of rows to plot
(in the specified sorting order) * alphabetical the merged
plot sorting order
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.
{kind=link}