
The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
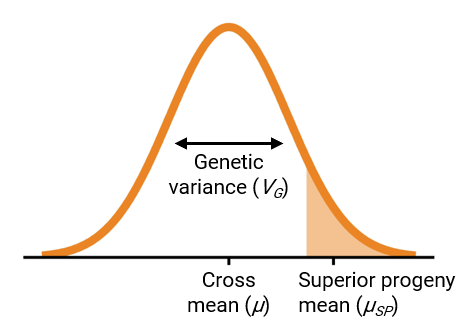
To make progress in breeding, populations should have a favorable mean and high genetic variance (Bernardo 2010). These two parameters can be combined into a single measure called the usefulness criterion (Schnell and Utz 1975), visualized in Figure 1.
Ideally, breeders would identify the set of parent combinations that,
when realized in a cross, would give rise to populations meeting these
requirements. PopVar is a package that uses phenotypic and
genomewide marker data on a set of candidate parents to predict the
mean, genetic variance, and superior progeny mean in bi-parental or
multi-parental populations. Thre package also contains functionality for
performing cross-validation to determine the suitability of different
statistical models. More details are available in Mohammadi, Tiede, and
Smith (2015) A dataset think_barley is included for
reference and examples.
You can install the released version of PopVar from CRAN with:
install.packages("PopVar")And the development version from GitHub with:
# install.packages("devtools")
devtools::install_github("UMN-BarleyOatSilphium/PopVar")Below is a description of the functions provided in
PopVar:
| Function | Description |
|---|---|
pop.predict |
Uses simulations to make predictions in recombinant inbred line populations; can internally perform cross-validation for model selections; can be quite slow. |
pop.predict2 |
Uses deterministic equations to make
predictions in populations of complete or partial selfing and with or
without the induction of doubled haploids; is much faster than
pop.predict; does not perform cross-validation or model
selection internally. |
pop_predict2 |
Has the same functionality as
pop.predict2, but accepts genomewide marker data in a
simpler matrix format. |
x.val |
Performs cross-validation to estimate model performance. |
mppop.predict |
Uses deterministic equations to make predictions in 2- or 4-way populations of complete or partial selfing and with or without the induction of doubled haploids; does not perform cross-validation or model selection internally. |
mpop_predict2 |
Has the same functionality as
mppop.predict, but accepts genomewide marker data in a
simpler matrix format. |
Examples are outlined in the package vignette.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.