The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
![]()
agriutilities is an R package designed to make the
analysis of field trials easier and more accessible for everyone working
in plant breeding. It provides a simple and intuitive interface for
conducting single and
multi-environmental trial analysis, with minimal coding
required. Whether you’re a beginner or an experienced user,
agriutilities will help you quickly and easily carry out complex
analyses with confidence. With built-in functions for fitting Linear
Mixed Models (LMM), agriutilities is the ideal choice
for anyone who wants to save time and focus on interpreting their
results.
install.packages("agriutilities")You can install the development version of agriutilities from GitHub with:
remotes::install_github("AparicioJohan/agriutilities")This is a basic example which shows you how to use some of the functions of the package.
The function check_design_met helps us to check the
quality of the data and also to identify the experimental design of the
trials. This works as a quality check or quality control before we fit
any model.
library(agriutilities)
library(agridat)
data(besag.met)
dat <- besag.met
results <- check_design_met(
data = dat,
genotype = "gen",
trial = "county",
traits = "yield",
rep = "rep",
block = "block",
col = "col",
row = "row"
)plot(results, type = "connectivity")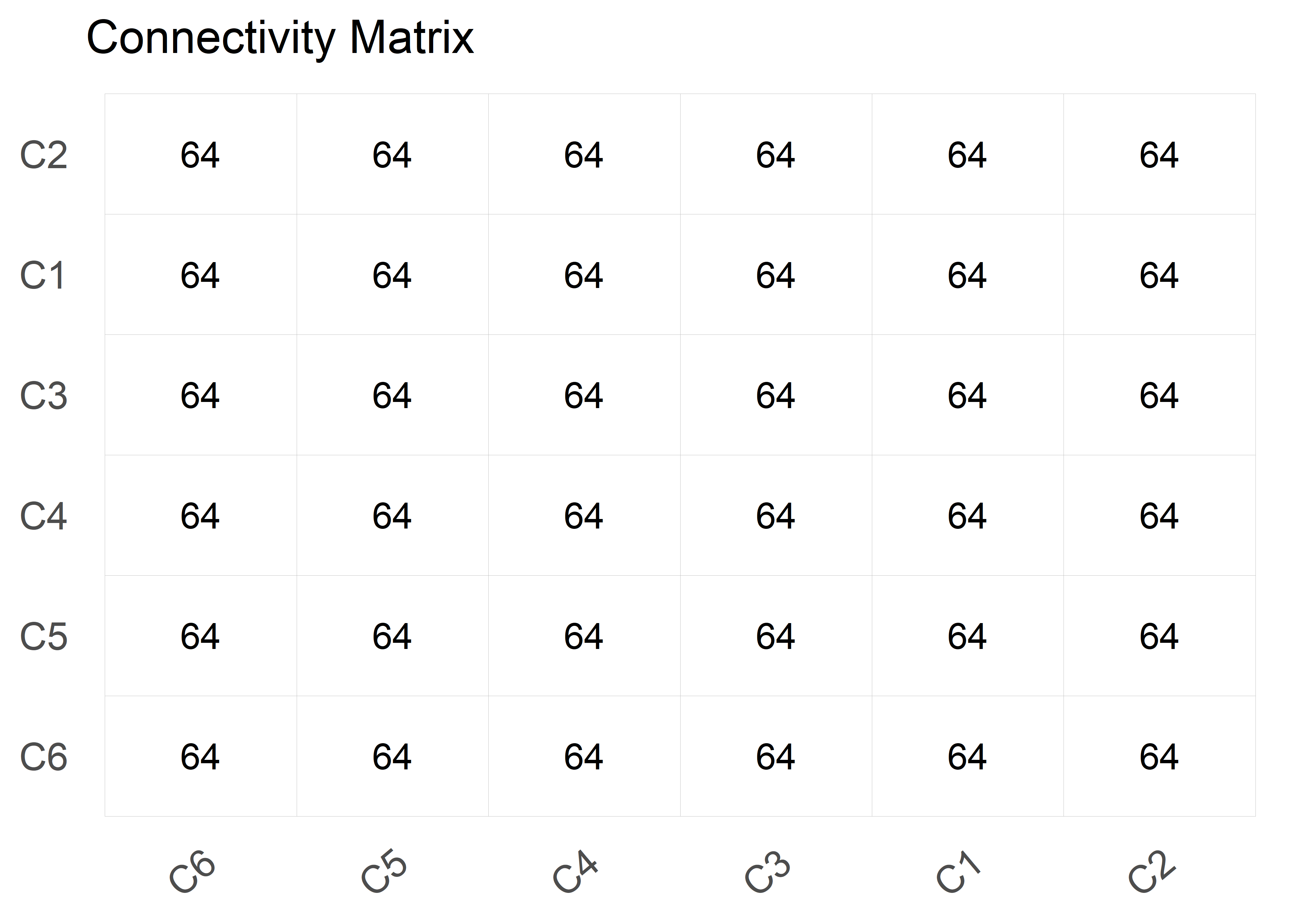
plot(results, type = "missing")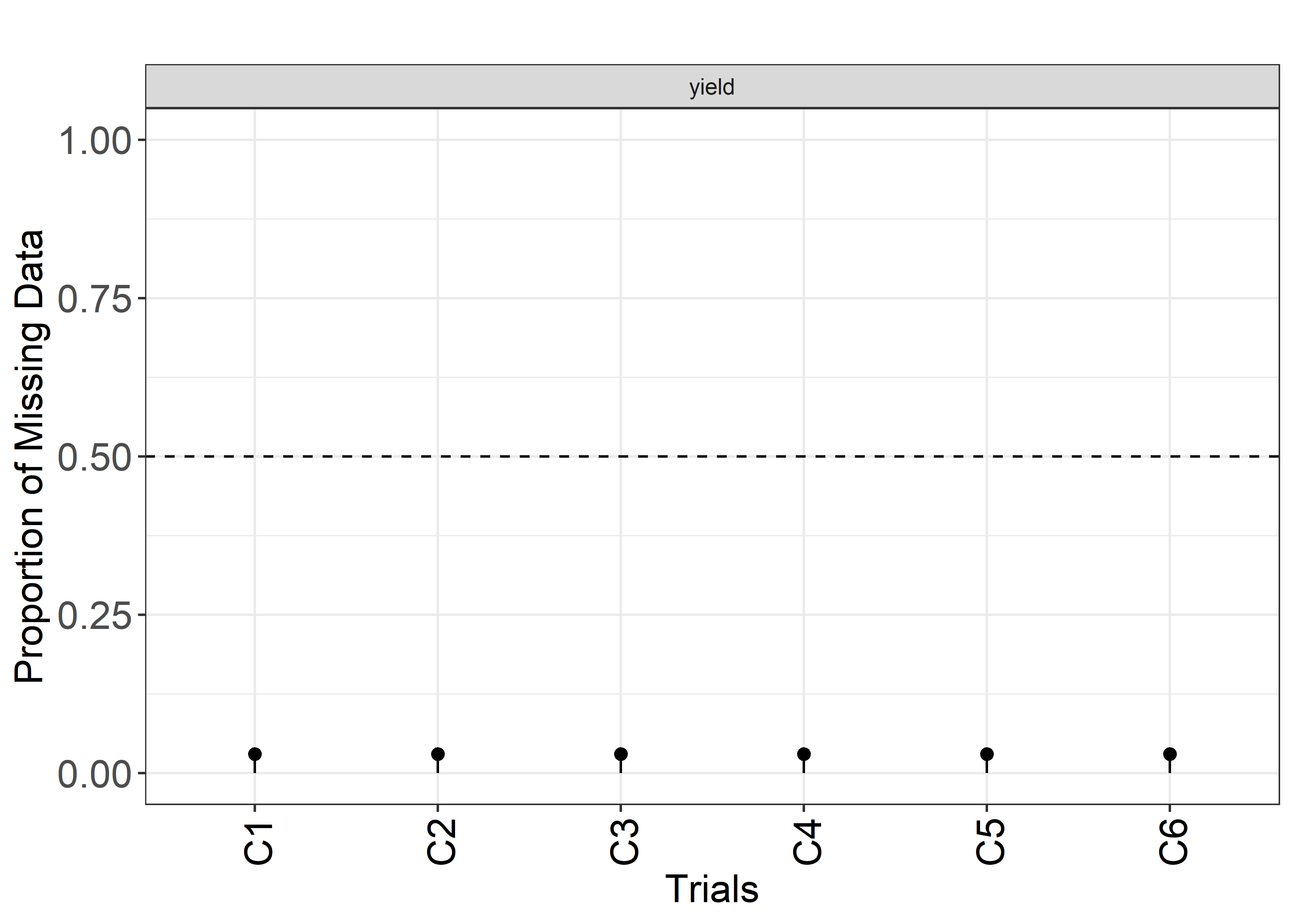
Inspecting the output.
print(results)
---------------------------------------------------------------------
Summary Traits by Trial:
---------------------------------------------------------------------
# A tibble: 6 × 11
county traits Min Mean Median Max SD CV n n_miss miss_perc
<fct> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <int> <int> <dbl>
1 C1 yield 87.9 149. 151. 200. 17.7 0.119 198 6 0.0303
2 C2 yield 24.4 56.1 52.1 125. 18.4 0.328 198 6 0.0303
3 C3 yield 28.2 87.9 89.2 137. 19.7 0.225 198 6 0.0303
4 C4 yield 103. 145. 143. 190. 17.1 0.118 198 6 0.0303
5 C5 yield 66.9 115. 116. 152. 16.4 0.142 198 6 0.0303
6 C6 yield 29.2 87.6 87.8 148. 26.6 0.304 198 6 0.0303
---------------------------------------------------------------------
Experimental Design Detected:
---------------------------------------------------------------------
county exp_design
1 C1 row_col
2 C2 row_col
3 C3 row_col
4 C4 row_col
5 C5 row_col
6 C6 row_col
---------------------------------------------------------------------
Summary Experimental Design:
---------------------------------------------------------------------
# A tibble: 6 × 9
county n n_gen n_rep n_block n_col n_row num_of_reps num_of_gen
<fct> <int> <int> <int> <int> <int> <int> <fct> <fct>
1 C1 198 64 3 8 11 18 3_9 63_1
2 C2 198 64 3 8 11 18 3_9 63_1
3 C3 198 64 3 8 11 18 3_9 63_1
4 C4 198 64 3 8 11 18 3_9 63_1
5 C5 198 64 3 8 11 18 3_9 63_1
6 C6 198 64 3 8 11 18 3_9 63_1
---------------------------------------------------------------------
Connectivity Matrix:
---------------------------------------------------------------------
C1 C2 C3 C4 C5 C6
C1 64 64 64 64 64 64
C2 64 64 64 64 64 64
C3 64 64 64 64 64 64
C4 64 64 64 64 64 64
C5 64 64 64 64 64 64
C6 64 64 64 64 64 64
---------------------------------------------------------------------
Filters Applied:
---------------------------------------------------------------------
List of 1
$ yield:List of 4
..$ missing_50% : chr(0)
..$ no_variation : chr(0)
..$ row_col_dup : chr(0)
..$ trials_to_remove: chr(0) The results of the previous function are used in
single_trial_analysis() to fit single trial models. This
function can fit, Completely Randomized Designs (CRD),
Randomized Complete Block Designs (RCBD), Resolvable
Incomplete Block Designs (res-IBD), Non-Resolvable
Row-Column Designs (Row-Col) and Resolvable Row-Column
Designs (res-Row-Col).
NOTE: It fits models based on the randomization detected.
obj <- single_trial_analysis(results, progress = FALSE)Inspecting the output.
print(obj)
---------------------------------------------------------------------
Summary Fitted Models:
---------------------------------------------------------------------
trait trial heritability CV VarGen VarErr design
<char> <char> <num> <num> <num> <num> <char>
1: yield C1 0.70 6.370054 85.28086 92.70982 row_col
2: yield C2 0.39 16.987235 26.87283 105.50494 row_col
3: yield C3 0.64 12.366843 82.84379 118.86865 row_col
4: yield C4 0.41 8.179794 35.75059 136.21686 row_col
5: yield C5 0.80 7.042116 104.44077 66.96454 row_col
6: yield C6 0.49 16.583972 72.16813 206.54020 row_col
---------------------------------------------------------------------
Outliers Removed:
---------------------------------------------------------------------
Null data.table (0 rows and 0 cols)
---------------------------------------------------------------------
First Predicted Values and Standard Errors (BLUEs/BLUPs):
---------------------------------------------------------------------
trait genotype trial BLUEs seBLUEs BLUPs seBLUPs wt
<char> <fctr> <fctr> <num> <num> <num> <num> <num>
1: yield G01 C1 142.9316 6.380244 144.5151 5.421481 0.02456549
2: yield G02 C1 156.7765 6.277083 155.0523 5.367425 0.02537957
3: yield G03 C1 126.5654 6.402526 133.1766 5.444349 0.02439480
4: yield G04 C1 155.7790 6.391590 154.2435 5.440070 0.02447836
5: yield G05 C1 163.9856 6.443261 160.7620 5.444314 0.02408732
6: yield G06 C1 129.5092 6.400364 134.7404 5.421543 0.02441129plot(obj, horizontal = TRUE, nudge_y_h2 = 0.12)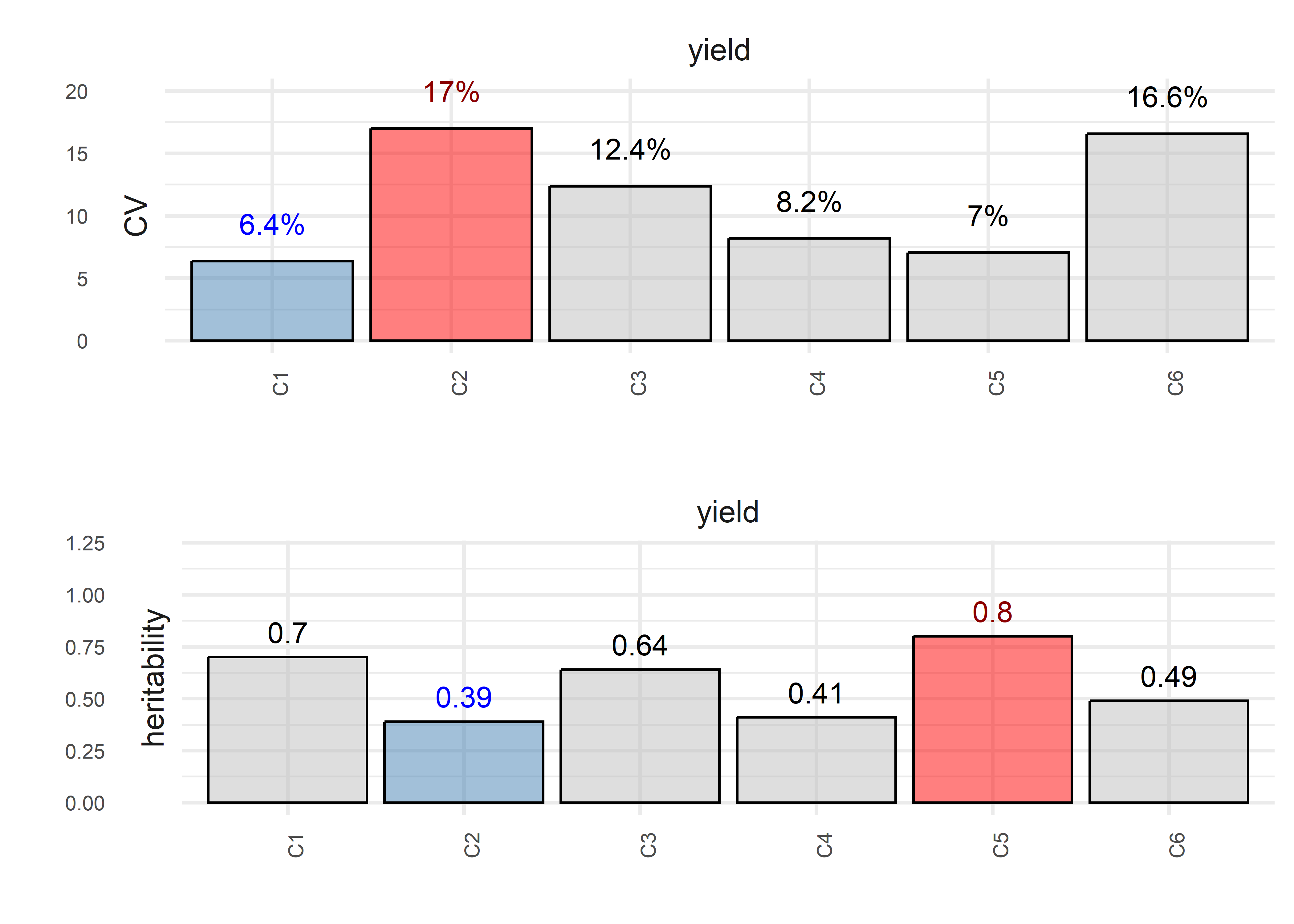
plot(obj, type = "correlation")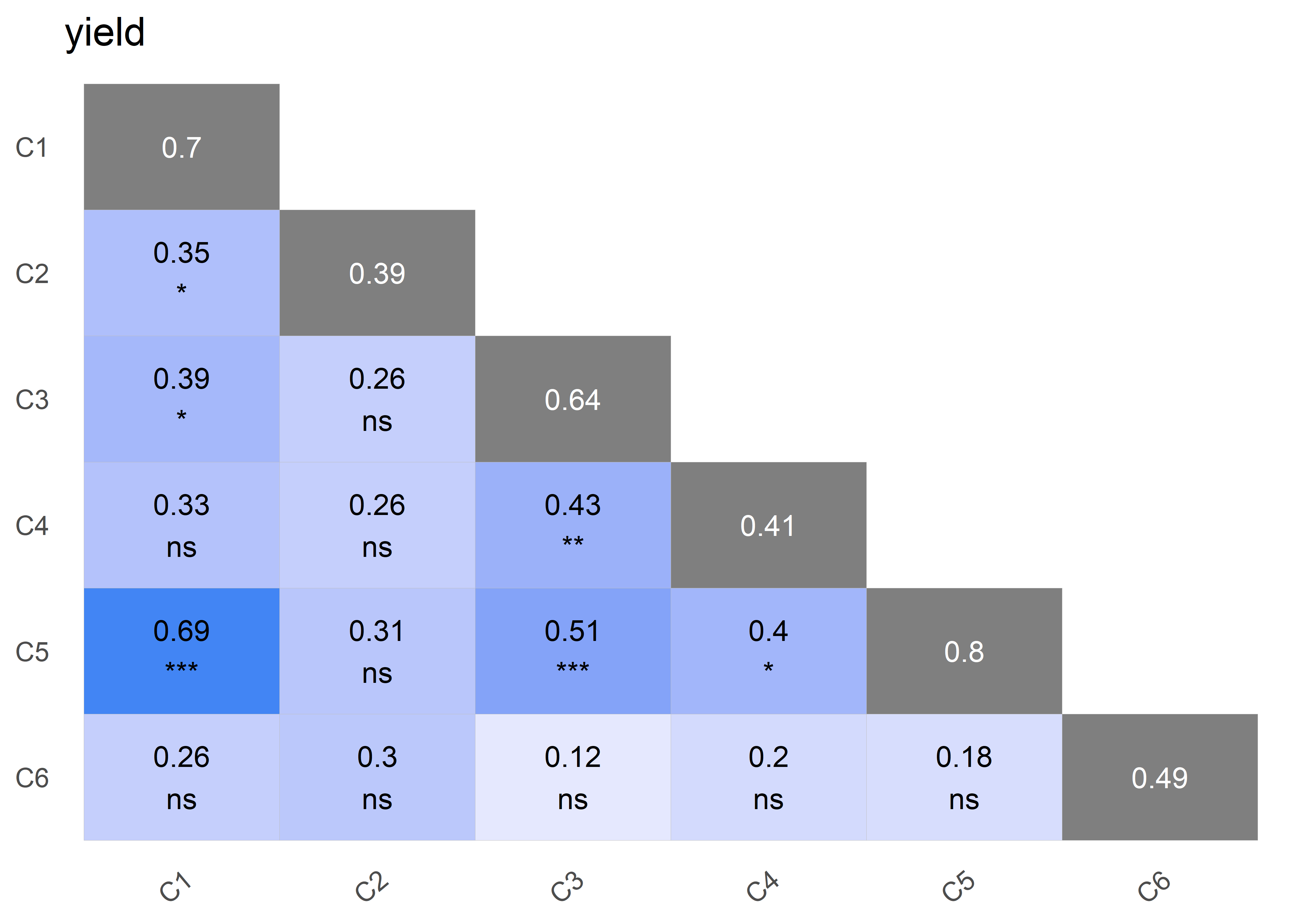
The returning object is a set of lists with trial summary, BLUEs, BLUPs, heritability, variance components, potential extreme observations, residuals, the models fitted and the data used.
The results of the previous function are used in
met_analysis() to fit multi-environmental trial models.
met_results <- met_analysis(obj, vcov = "fa2", progress = FALSE)
Online License checked out Thu Oct 16 21:37:04 2025Inspecting the output.
print(met_results)
---------------------------------------------------------------------
Trial Effects (BLUEs):
---------------------------------------------------------------------
trait trial predicted.value std.error status
1 yield C1 149.59014 1.369766 Estimable
2 yield C2 67.20545 1.137972 Estimable
3 yield C3 90.79958 1.441812 Estimable
4 yield C4 148.12623 1.172130 Estimable
5 yield C5 122.40195 1.440843 Estimable
6 yield C6 88.35437 1.530165 Estimable
---------------------------------------------------------------------
Heritability:
---------------------------------------------------------------------
trait h2
1 yield 0.8261367
---------------------------------------------------------------------
First Overall Predicted Values and Standard Errors (BLUPs):
---------------------------------------------------------------------
trait genotype predicted.value std.error status
1 yield G01 110.8429 2.536428 Estimable
2 yield G02 111.3836 2.548777 Estimable
3 yield G03 102.6612 2.551662 Estimable
4 yield G04 115.8775 2.546016 Estimable
5 yield G05 121.0640 2.558195 Estimable
6 yield G06 108.9498 2.570048 Estimable
---------------------------------------------------------------------
Variance-Covariance Matrix:
---------------------------------------------------------------------
Correlation Matrix ('fa2'): yield
C1 C2 C3 C4 C5 C6
C1 1.00 0.64 0.71 0.86 0.95 0.42
C2 0.64 1.00 0.58 0.86 0.53 0.70
C3 0.71 0.58 1.00 0.70 0.71 0.39
C4 0.86 0.86 0.70 1.00 0.83 0.58
C5 0.95 0.53 0.71 0.83 1.00 0.34
C6 0.42 0.70 0.39 0.58 0.34 1.00
Covariance Matrix ('fa2'): yield
C1 C2 C3 C4 C5 C6
C1 78.93 29.35 55.78 38.05 85.20 30.76
C2 29.35 26.42 26.61 21.93 27.81 29.68
C3 55.78 26.61 78.65 30.97 63.27 28.46
C4 38.05 21.93 30.97 24.55 41.49 23.89
C5 85.20 27.81 63.27 41.49 102.30 28.28
C6 30.76 29.68 28.46 23.89 28.28 67.99
---------------------------------------------------------------------
First Stability Coefficients:
---------------------------------------------------------------------
trait genotype superiority static wricke predicted.value
1 yield G57 22.64170 32.92556 15.484987 92.63362
2 yield G29 17.03322 33.66855 5.023783 99.67843
3 yield G34 17.02203 33.06040 8.545979 100.06674
4 yield G59 16.72402 34.06416 5.596864 100.14511
5 yield G31 15.77027 31.36932 10.740548 102.04249
6 yield G10 15.59219 32.03990 11.767180 102.64704pvals <- met_results$trial_effects
model <- met_results$met_models$yield
fa_objt <- fa_summary(
model = model,
trial = "trial",
genotype = "genotype",
BLUEs_trial = pvals,
k_biplot = 8,
size_label_var = 4,
filter_score = 1
)fa_objt$plots$loadings_c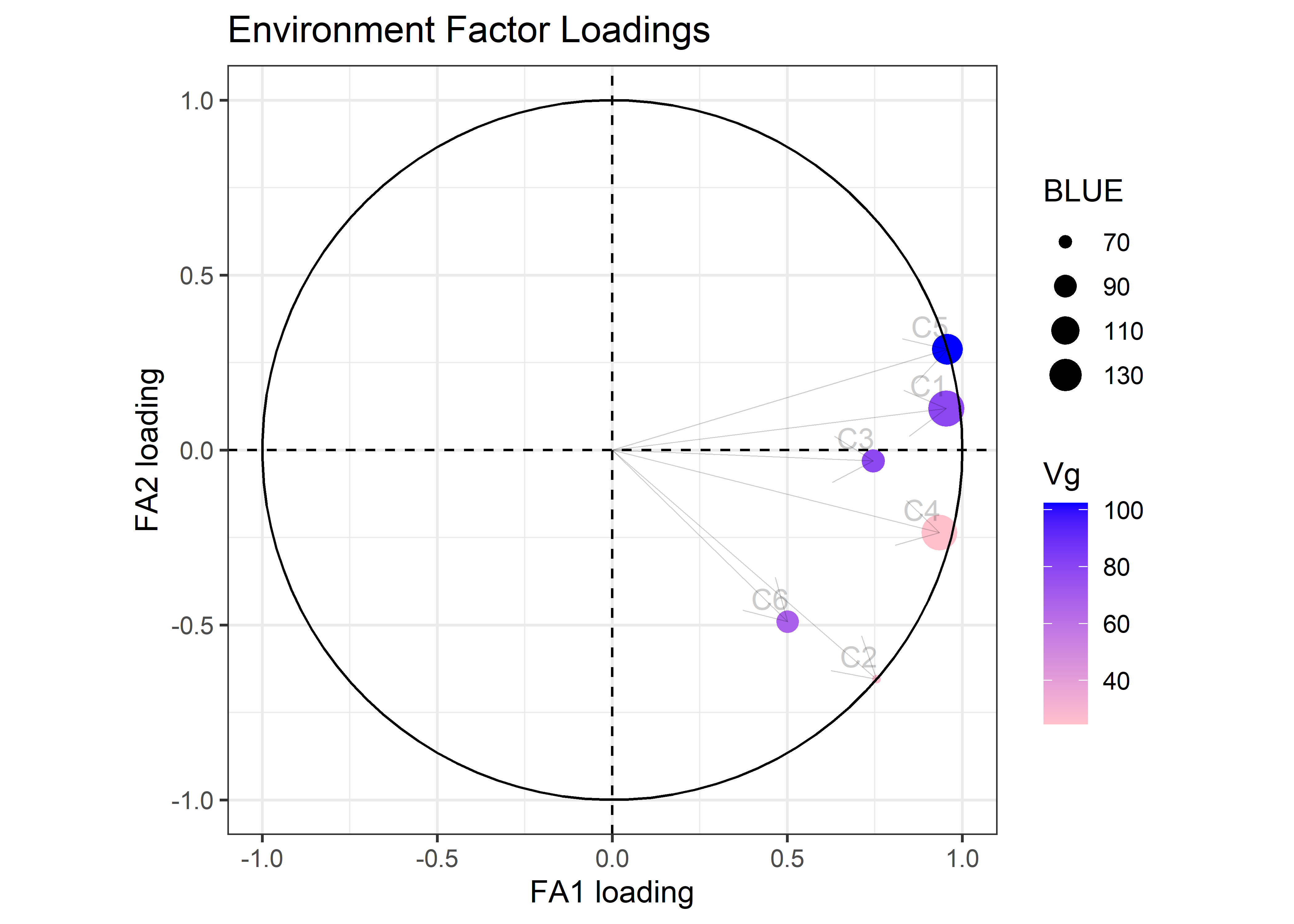
fa_objt$plots$biplot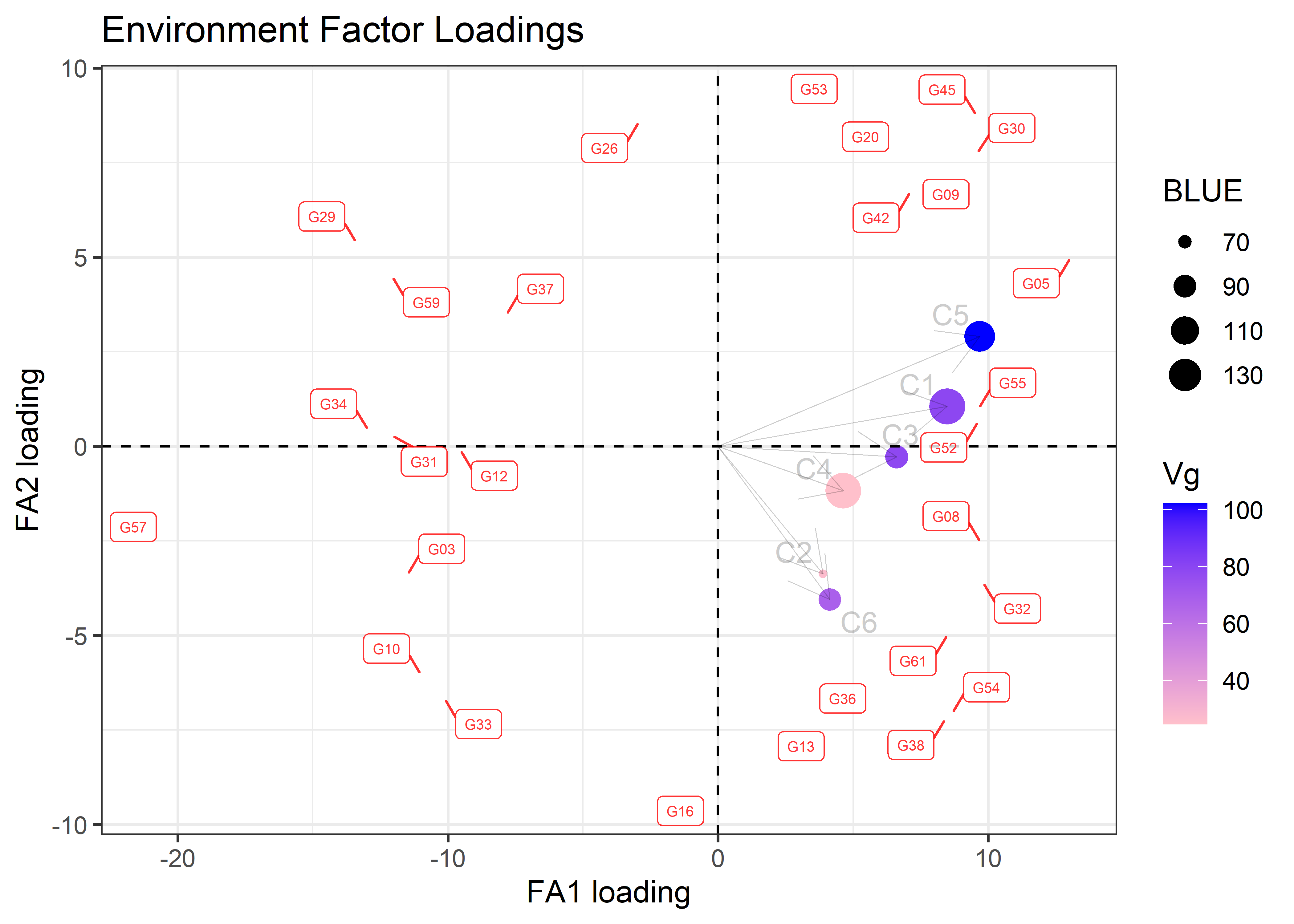
For more information and to learn more about what is described here you may find useful the following sources: Isik, Holland, and Maltecca (2017); Rodriguez-Alvarez et al. (2018).
Please note that the agriutilities project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.