distantia Time
Series Dissimilarity
The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
distantia Time
Series Dissimilarity
![]()
![]()
The R package distantia offers an
efficient, feature-rich toolkit for managing, comparing, and analyzing
time series data. It is designed to handle a wide range of scenarios,
including:
distantia::distances.psi.zoo objects.tsl_...() functions for generating,
resampling, transforming, analyzing, and visualizing univariate and
multivariate time series.If you find this package useful, please cite it as:
Blas M. Benito, H. John B. Birks (2020). distantia: an open-source toolset to quantify dissimilarity between multivariate ecological time-series. Ecography, 43(5), 660-667. doi: 10.1111/ecog.04895.
Blas M. Benito (2024). distantia: A Toolset for Time Series Dissimilarity Analysis. R package version 2.0. url: https://blasbenito.github.io/distantia/.
Version 2.0.0 of distantia can be installed from CRAN
and GitHub.
#CRAN
install.packages("distantia")
#GitHub
remotes::install_github(
repo = "blasbenito/distantia",
ref = "main"
)This section showcases several features of the package
distantia. Please check the Articles
section for further details.
All heavy-duty functions in distantia support
parallelization via the future package. However, due
to the high efficiency of the C++ backend of distantia,
parallel execution is only worth it for very large datasets and
restricted permutation analyses.
Progress bars provided by the progressr package are also available. Unfortunately, the latter does not work in R Markdown documents like this one.
library(distantia, quietly = TRUE)
library(future)
# library(progressr)
#parallelization setup
# future::plan(future::multisession)
#progress bar (does not work in Rmarkdown)
#progressr::handlers(global = TRUE)The albatross data frame contains daily GPS data of 4
individuals of Waved Albatross in the Pacific captured during the summer
of 2008. Below are the first 6 rows of this data frame:
#> name time x y speed temperature heading
#> 1 X132 2008-05-31 -89.62097 -1.389512 0.1473333 29.06667 212.0307
#> 2 X132 2008-06-01 -89.62101 -1.389508 0.2156250 28.25000 184.0337
#> 3 X132 2008-06-02 -89.62101 -1.389503 0.2143750 27.68750 123.1269
#> 4 X132 2008-06-03 -89.62099 -1.389508 0.2018750 27.81250 183.4600
#> 5 X132 2008-06-04 -89.62098 -1.389507 0.2256250 27.68750 114.8931
#> 6 X132 2008-06-05 -89.62925 -1.425734 1.3706667 25.73333 245.8033The code below transforms the data to a Time Series List
with tsl_initialize() and applies global scaling and
centering with tsl_transform() and
f_scale_global to facilitate time series comparisons.
tsl <- tsl_initialize(
x = distantia::albatross,
name_column = "name",
time_column = "time",
lock_step = TRUE
) |>
tsl_transform(
f = f_scale_global
)
tsl_plot(
tsl = tsl,
ylim = "relative"
)
Lock-step analysis performs direct comparisons between samples observed at the same time without any time distortion. It requires time series of the same length, preferably observed at the same times.
df_ls <- distantia(
tsl = tsl,
lock_step = TRUE
)
df_ls[, c("x", "y", "psi")]
#> x y psi
#> 1 X132 X134 1.888451
#> 3 X132 X153 2.128340
#> 5 X134 X153 2.187862
#> 4 X134 X136 2.270977
#> 2 X132 X136 2.427479
#> 6 X136 X153 2.666099The “psi” column shows normalized dissimilarity values and is used to sort the data frame from lowest to highest dissimilarity. Hence, the first row shows the most similar pair of time series.
The function distantia_boxplot() enables a quick
identification of the time series that are either more dissimilar (top)
or similar (bottom) to others.
distantia_boxplot(df = df_ls, text_cex = 0.8)
By default, distantia() computes unrestricted dynamic
time warping with orthogonal and diagonal least-cost paths.
df_dtw <- distantia(
tsl = tsl
)
df_dtw[, c("x", "y", "psi")]
#> x y psi
#> 1 X132 X134 1.163499
#> 5 X134 X153 2.115504
#> 4 X134 X136 2.206298
#> 3 X132 X153 2.306471
#> 2 X132 X136 2.307844
#> 6 X136 X153 2.686551The function distantia_dtw_plot() provides detailed
insights into the alignment between a pair of time series resulting from
DTW.
distantia_dtw_plot(
tsl = tsl[c("X132", "X153")]
)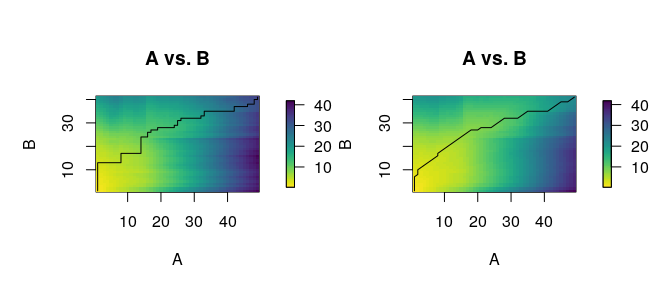
Deviations from the perfect diagonal in the least-cost path reveal adjustments made by DTW to align time series by shape rather than time.
The article Dynamic Time Warping vs Lock-Step provides further insights into the advantages and disadvantages of each method in different scenarios.
The function distantia() implements restricted
permutation tests to assess the significance of dissimilarity scores. It
provides several setups to support different assumptions.
For example, the configuration below rearranges complete rows within 7-day blocks, assuming strong dependencies within rows and between observations that are close in time.
future::plan(future::multisession)
df_dtw <- distantia(
tsl = tsl,
repetitions = 1000,
permutation = "restricted_by_row",
block_size = 7 #one week
)
future::plan(future::sequential)
df_dtw[, c("x", "y", "psi", "p_value")]
#> x y psi p_value
#> 1 X132 X134 1.163499 0.001
#> 5 X134 X153 2.115504 0.380
#> 4 X134 X136 2.206298 0.134
#> 3 X132 X153 2.306471 0.376
#> 2 X132 X136 2.307844 0.002
#> 6 X136 X153 2.686551 0.005The “p_value” column represents the fraction of permutations yielding a psi score lower than the observed value, and indicates the strength of similarity between two time series. A significance threshold (e.g., 0.05, depending on the number of repetitions) helps identify pairs of time series with a robust similarity.
When comparing multivariate time series, certain variables contribute
more to similarity or dissimilarity. The momentum()
function uses a leave-one-out algorithm to quantify each variable’s
contribution to the overall dissimilarity between two time series.
df_importance <- momentum(
tsl = tsl
)
df_importance[, c("x", "y", "variable", "importance", "effect")]
#> x y variable importance effect
#> 1 X132 X134 x 89.721187 decreases similarity
#> 2 X132 X134 y 101.305396 decreases similarity
#> 3 X132 X134 speed -28.286386 increases similarity
#> 4 X132 X134 temperature 78.130508 decreases similarity
#> 5 X132 X134 heading -43.644053 increases similarity
#> 6 X132 X136 x 15.687570 decreases similarity
#> 7 X132 X136 y 82.867368 decreases similarity
#> 8 X132 X136 speed -67.196851 increases similarity
#> 9 X132 X136 temperature 382.039900 decreases similarity
#> 10 X132 X136 heading -104.245839 increases similarity
#> 11 X132 X153 x 467.261463 decreases similarity
#> 12 X132 X153 y 159.727491 decreases similarity
#> 13 X132 X153 speed -44.549191 increases similarity
#> 14 X132 X153 temperature -4.016121 increases similarity
#> 15 X132 X153 heading -88.852346 increases similarity
#> 16 X134 X136 x 36.205194 decreases similarity
#> 17 X134 X136 y 90.757712 decreases similarity
#> 18 X134 X136 speed -61.923595 increases similarity
#> 19 X134 X136 temperature 348.244258 decreases similarity
#> 20 X134 X136 heading -96.737145 increases similarity
#> 21 X134 X153 x 761.132445 decreases similarity
#> 22 X134 X153 y 26.329542 decreases similarity
#> 23 X134 X153 speed -62.801312 increases similarity
#> 24 X134 X153 temperature -23.264433 increases similarity
#> 25 X134 X153 heading -76.874072 increases similarity
#> 26 X136 X153 x 530.402462 decreases similarity
#> 27 X136 X153 y 24.217594 decreases similarity
#> 28 X136 X153 speed -67.659029 increases similarity
#> 29 X136 X153 temperature 255.711643 decreases similarity
#> 30 X136 X153 heading -119.877355 increases similarityPositive “importance” values indicate variables contributing to dissimilarity, while negative values indicate contributions to similarity. The function documentation provides more details on how importance scores are computed.
The momentum_boxplot() function provides a quick insight
into which variables contribute the most to similarity or dissimilarity
across all pairs of time series.
momentum_boxplot(
df = df_importance
)
The package distantia provides tools to group time
series by dissimilarity using hierarchical or K-means clustering. The
example below applies the former to the albatross dataset
to identify groups of individuals with the most similar movement time
series.
dtw_hclust <- distantia_cluster_hclust(
df = df_dtw,
clusters = NULL, #automatic mode
method = NULL #automatic mode
)
#cluster object
dtw_hclust$cluster_object
#>
#> Call:
#> stats::hclust(d = d_dist, method = method)
#>
#> Cluster method : ward.D
#> Number of objects: 4
#number of clusters
dtw_hclust$clusters
#> [1] 3
#clustering data frame
#group label in column "cluster"
# negatives in column "silhouette_width" highlight anomalous cluster assignments
dtw_hclust$df
#> name cluster silhouette_width
#> 1 X132 1 0.4955501
#> 2 X134 1 0.4500134
#> 3 X136 2 0.0000000
#> 4 X153 3 0.0000000
#tree plot
par(mar=c(3,1,1,3))
plot(
x = stats::as.dendrogram(
dtw_hclust$cluster_object
),
horiz = TRUE
)
This is just a summary of the features implemented in the package.
Please visit the Articles section to find out more
about distantia.
If you encounter bugs or issues with the documentation, please file an issue on GitHub.
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.