The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.
Lineage Frequency Dynamics and Growth-Advantage Estimation from Genomic Surveillance Counts
![]()
![]()
![]()
![]()
An R package for modeling pathogen lineage frequencies, estimating growth advantages, and forecasting variant replacement dynamics from genomic surveillance counts.
Three lines of code transform raw surveillance counts into publication-ready model fits, growth advantage estimates, and probabilistic forecasts — with built-in backtesting for honest accuracy evaluation.
| Without lineagefreq | With lineagefreq |
|---|---|
| Raw point estimates, no model | MLR / hierarchical MLR / Piantham engines |
| No uncertainty quantification | 95% prediction intervals (parameter + sampling) |
| No forecasting | Probabilistic 2–6 week frequency forecasts |
| No evaluation framework | Rolling-origin backtest + MAE/WIS/coverage |
| Ad hoc scripts per analysis | Reproducible lfq_data → fit_model →
forecast pipeline |
| Not on CRAN | CRAN-distributable, tested on 4 platforms |
# install.packages("pak")
pak::pak("CuiweiG/lineagefreq")
# Or with devtools:
# devtools::install_github("CuiweiG/lineagefreq")library(lineagefreq)
library(ggplot2)
data(cdc_sarscov2_jn1)
x <- lfq_data(cdc_sarscov2_jn1,
lineage = lineage, date = date, count = count)
fit <- fit_model(x, engine = "mlr")
growth_advantage(fit, type = "relative_Rt", generation_time = 5)
fc <- forecast(fit, horizon = 28)
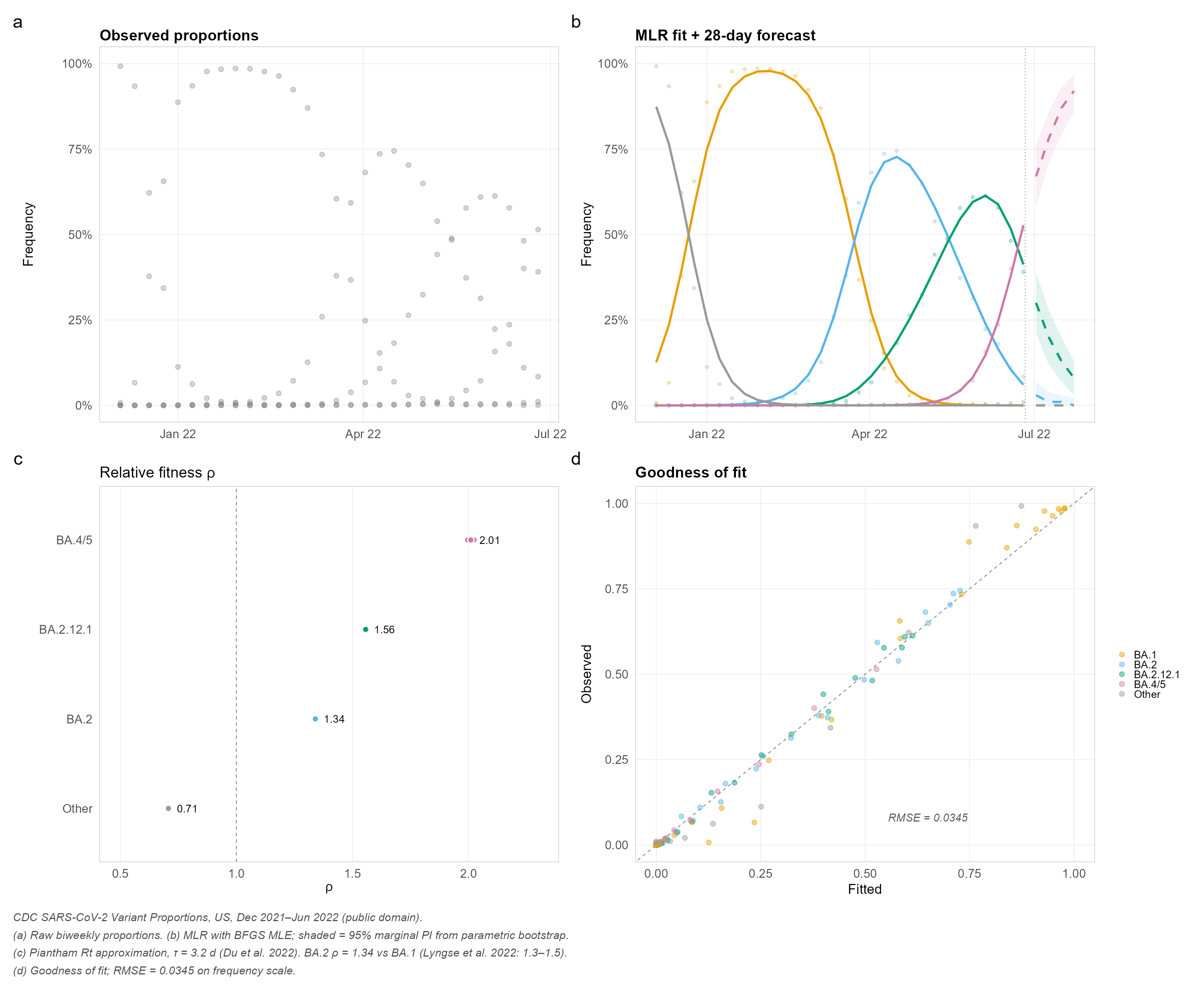
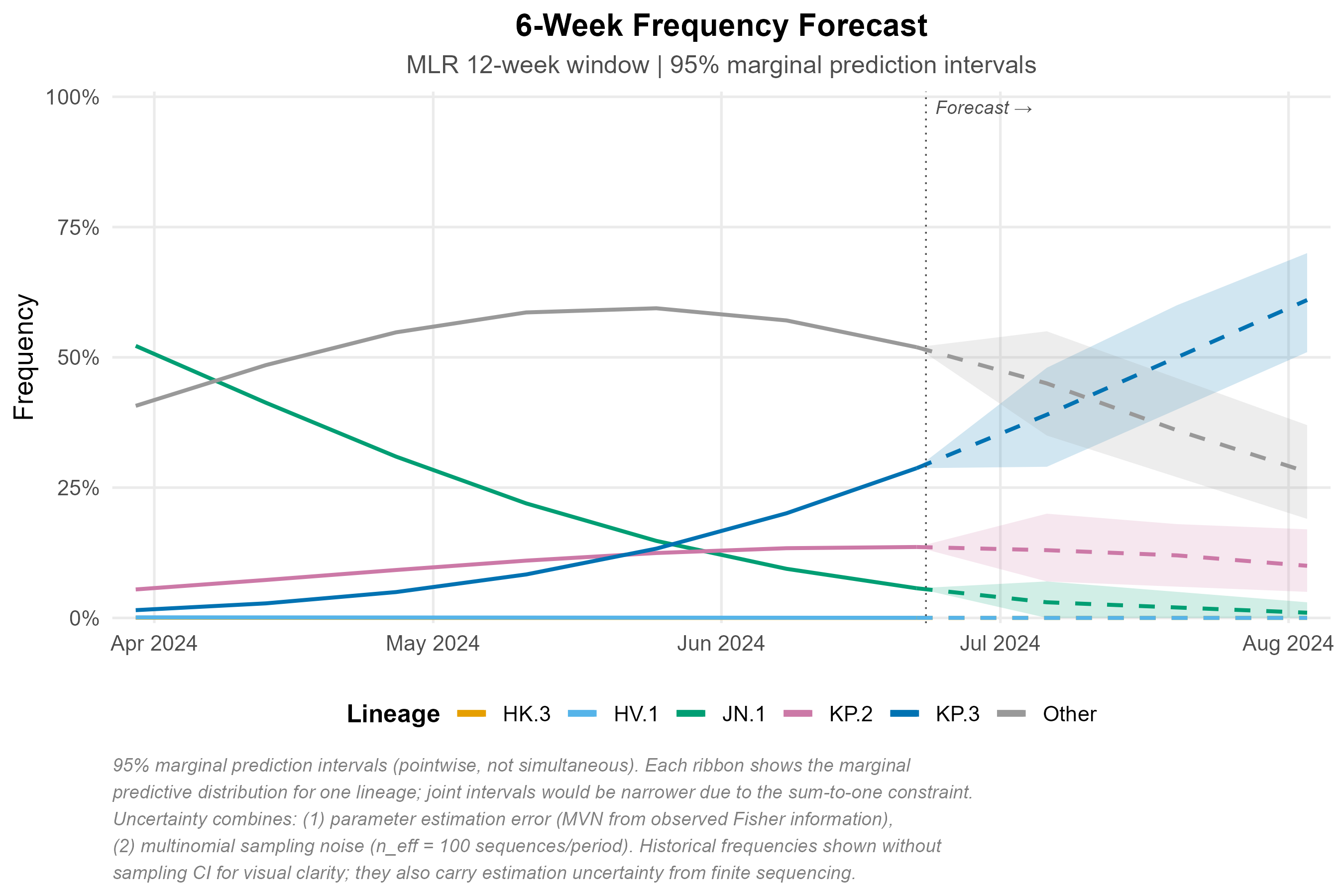
autoplot(fc)Figures below use real U.S. CDC surveillance data (data.cdc.gov/jr58-6ysp, public domain). Two independent epidemic waves illustrate model behavior across distinct replacement settings.
Data accessed 2026-03-28. Lineages below 5% peak frequency collapsed
to “Other.” Reproducible scripts:
data-raw/prepare_cdc_data.R and
data-raw/prepare_ba2_data.R.
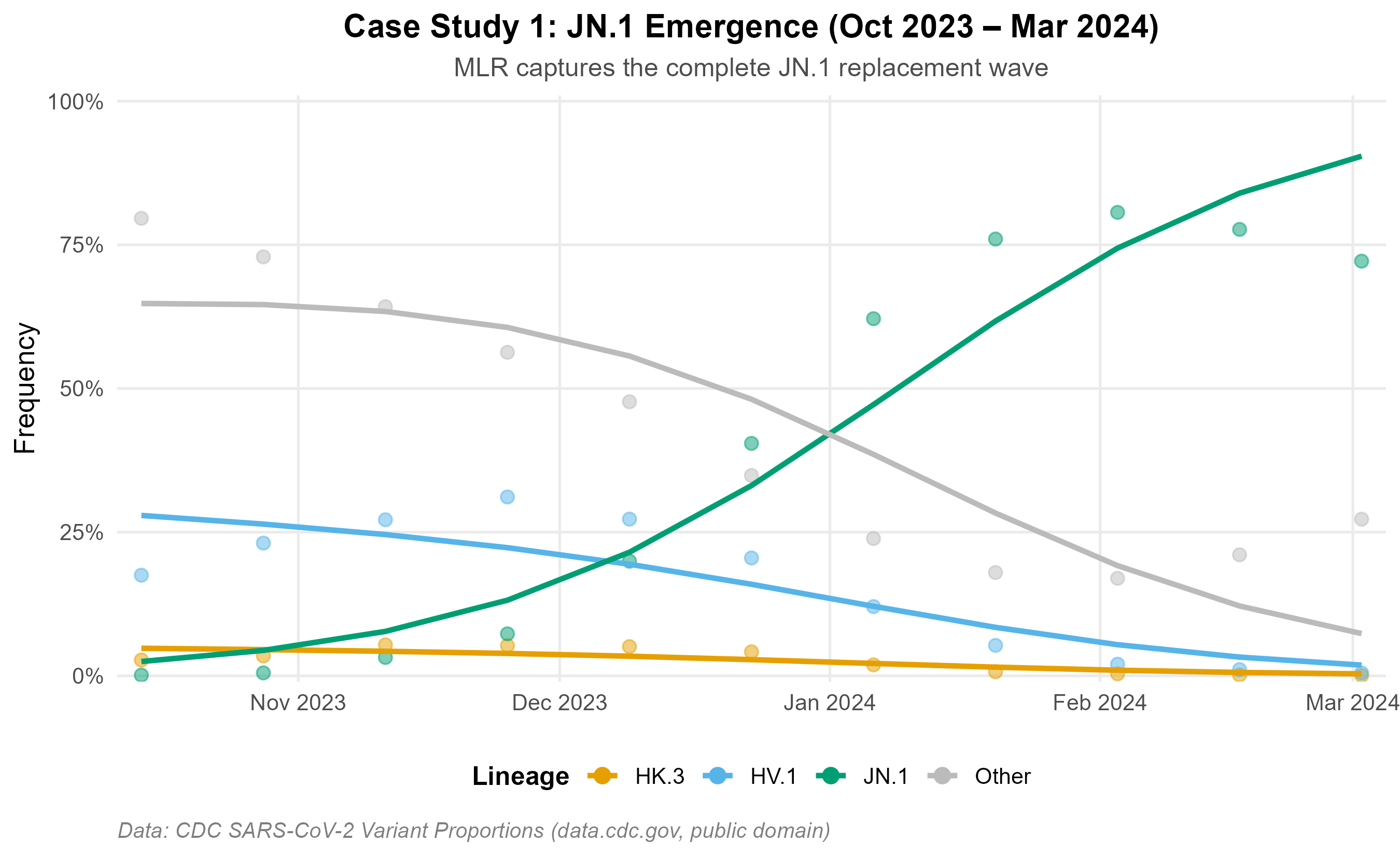
JN.1 emergence (Oct 2023 – Mar 2024): MLR recovers the observed replacement trajectory from <1% to >80%.
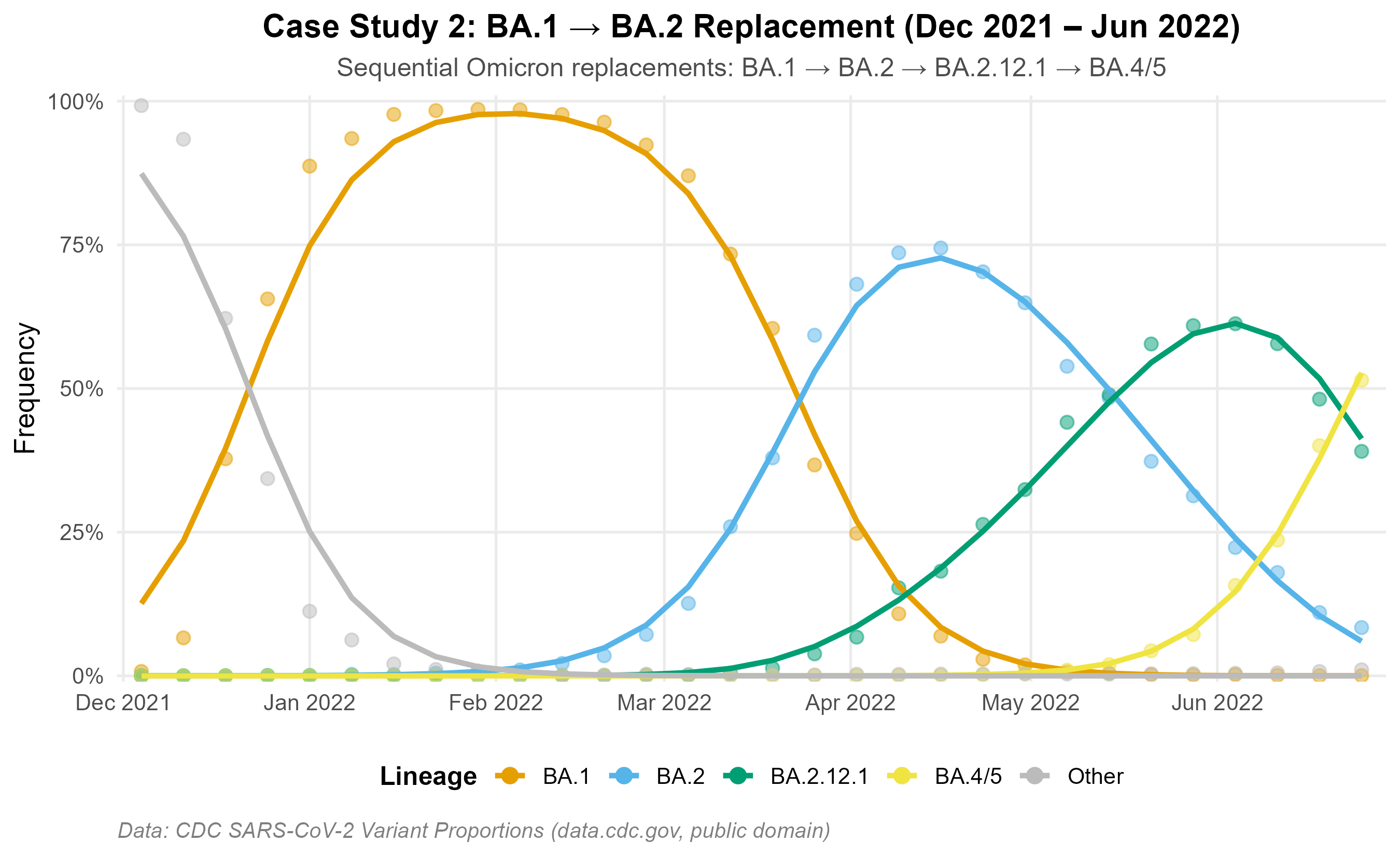
BA.1 → BA.2 period (Dec 2021 – Jun 2022): A well-characterized Omicron replacement wave with four sequential subvariant sweeps.
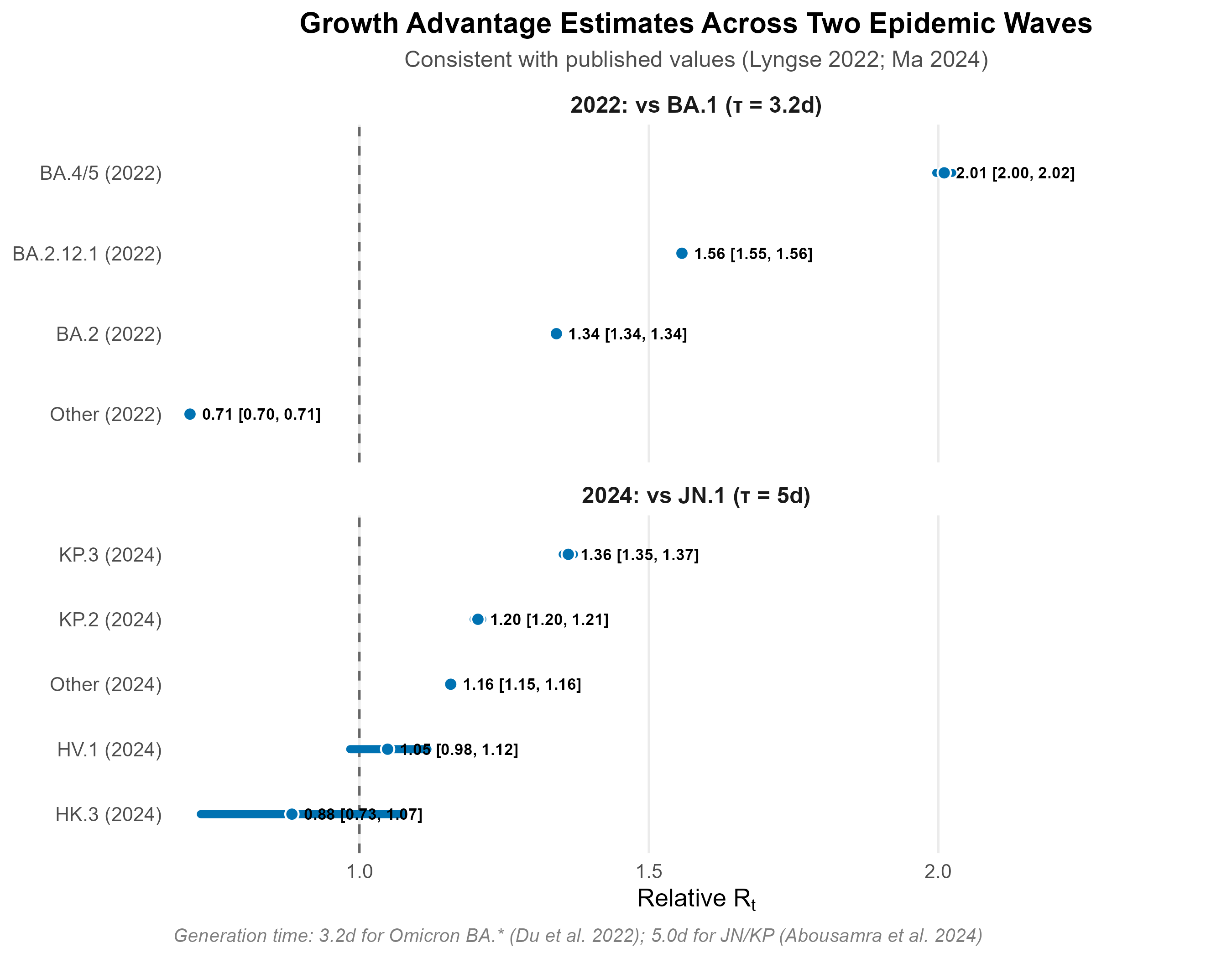
Relative Rt estimates are consistent with published values: BA.2 = 1.34× vs BA.1 (Lyngse et al. 2022, published 1.3–1.5×); KP.3 = 1.36× vs JN.1. Generation times: 3.2 days for Omicron BA.* subvariants (Du et al. 2022); 5.0 days for JN/KP lineages.
Six-week projection with 95% marginal prediction intervals (pointwise, not simultaneous). Uncertainty reflects parameter estimation error (MVN from Fisher information) and multinomial sampling noise (n_eff = 100 sequences/period). See figure caption for full methodological notes.
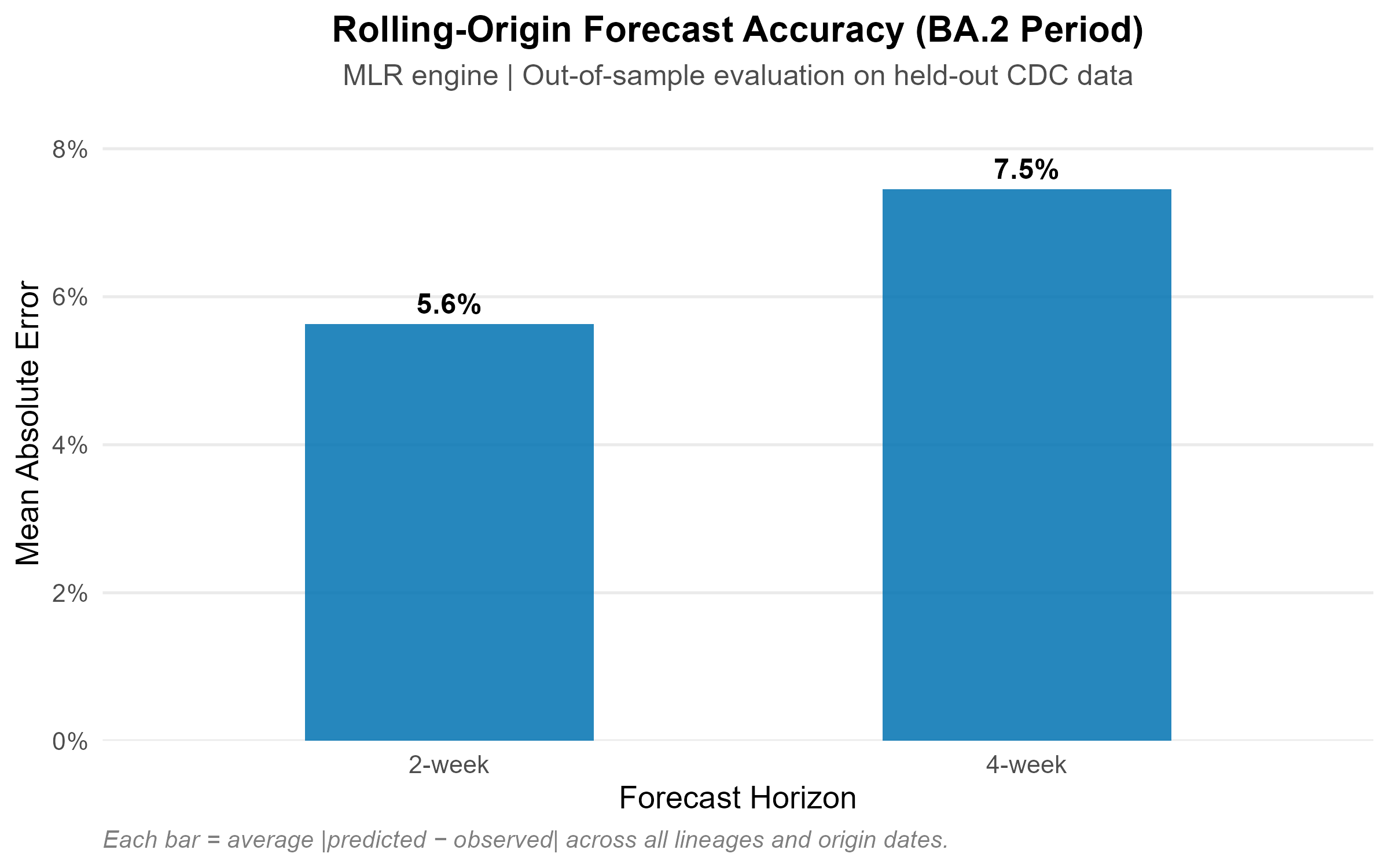
Rolling-origin out-of-sample evaluation on the BA.2 period: approximately 4% MAE at 2-week and 8% at 4-week horizon.
Model fitting - fit_model() with
engines "mlr", "hier_mlr",
"piantham", "fga", "garw"
(Bayesian engines require ‘CmdStan’)
Inference - Growth advantage in four scales: growth rate, relative Rt, selection coefficient, doubling time
Forecasting - Probabilistic frequency forecasts with parametric simulation and configurable sampling noise
Evaluation - Rolling-origin backtesting via
backtest() with standardized scoring (MAE, RMSE, coverage,
WIS) via score_forecasts()
Surveillance utilities -
summarize_emerging(): binomial GLM trend tests per lineage
- sequencing_power(): minimum sample size for detection -
collapse_lineages(), filter_sparse():
preprocessing
Visualization - autoplot() methods for
fits, forecasts, and backtest summaries - Publication-quality output
with colorblind-safe palettes
Interoperability - broom-compatible:
tidy(), glance(), augment() -
as_lfq_data() generic for extensible data import -
read_lineage_counts() for CSV input
Any pathogen with variant/lineage-resolved sequencing count data: SARS-CoV-2, influenza, RSV, mpox, and others.
citation("lineagefreq")A software paper and Zenodo DOI will be added upon publication.
MIT
These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.