The hardware and bandwidth for this mirror is donated by METANET, the Webhosting and Full Service-Cloud Provider.
If you wish to report a bug, or if you are interested in having us mirror your free-software or open-source project, please feel free to contact us at mirror[@]metanet.ch.



rbiom is designed for microbiome researchers, providing
visualizations and statistical analyses from Biological Observation
Matrix (BIOM) files.
The latest stable version can be installed from CRAN.
install.packages('rbiom')The development version is available on GitHub.
install.packages('pak')
pak::pak('cmmr/rbiom')library(rbiom)
infile <- system.file(package = 'rbiom', 'extdata', 'hmp50.bz2')
biom <- rarefy(infile)bdiv_ord_plot(biom, stat.by = 'Body Site', facet.by = 'Sex')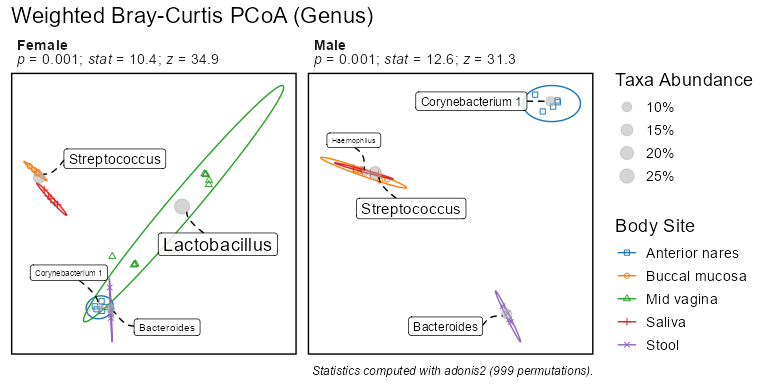
adiv_boxplot(biom, x = 'Sex', adiv = c('otu', 'shan'), stat.by = 'Body Site')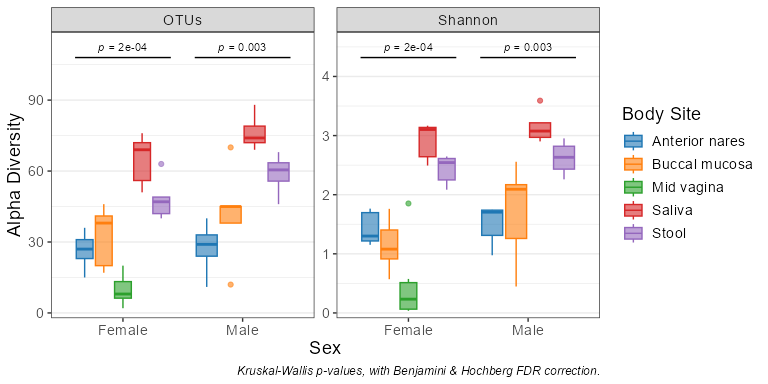
subset(biom, `Body Site` == 'Buccal mucosa') %>%
taxa_corrplot('Age', taxa = 2, layers = 'ptc', fit = 'lm', test = 'emtrends')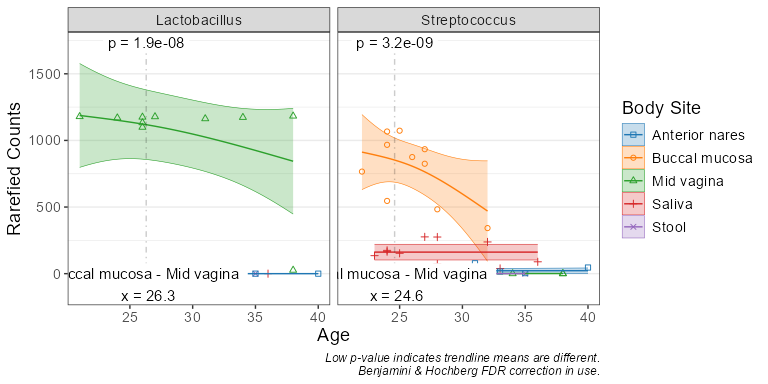
taxa_heatmap(biom, taxa = 10, tracks = c('body', 'age'))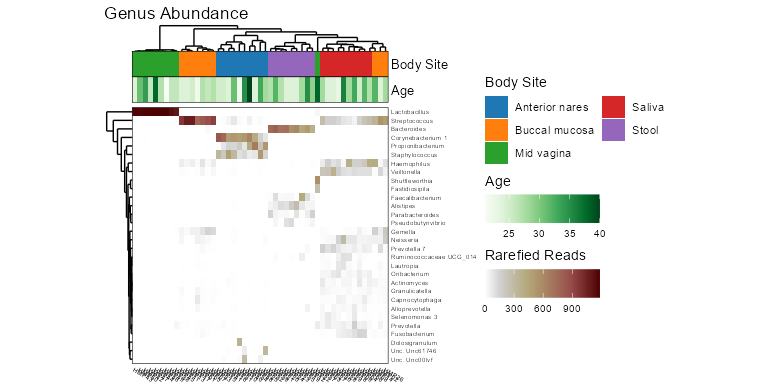
taxa_stacked(biom, rank = 'Phylum')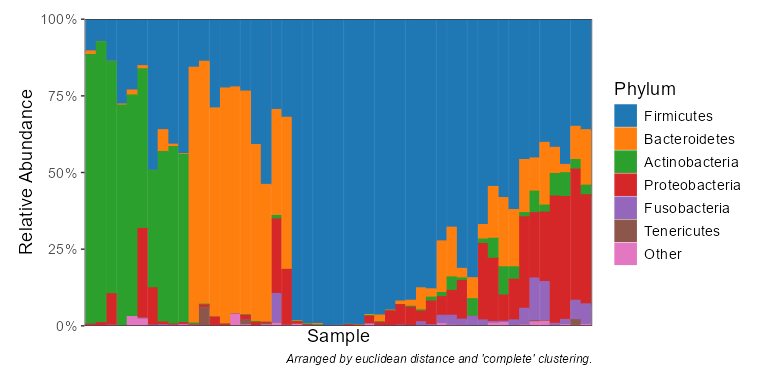
taxa_table(biom, 'Phylum')
#> # A tibble: 294 × 8
#> .rank .sample .taxa .abundance Age BMI `Body Site` Sex
#> <fct> <chr> <fct> <dbl> <dbl> <dbl> <fct> <fct>
#> 1 Phylum HMP01 Firmicutes 866 22 20 Buccal mucosa Female
#> 2 Phylum HMP01 Bacteroidetes 202 22 20 Buccal mucosa Female
#> 3 Phylum HMP01 Actinobacteria 14 22 20 Buccal mucosa Female
#> 4 Phylum HMP01 Proteobacteria 60 22 20 Buccal mucosa Female
#> 5 Phylum HMP01 Fusobacteria 33 22 20 Buccal mucosa Female
#> 6 Phylum HMP01 Tenericutes 0 22 20 Buccal mucosa Female
#> 7 Phylum HMP02 Firmicutes 814 24 23 Buccal mucosa Male
#> 8 Phylum HMP02 Bacteroidetes 181 24 23 Buccal mucosa Male
#> 9 Phylum HMP02 Actinobacteria 49 24 23 Buccal mucosa Male
#> 10 Phylum HMP02 Proteobacteria 97 24 23 Buccal mucosa Male
#> # ℹ 284 more rowsThe online manual for rbiom is available at https://cmmr.github.io/rbiom/. It includes a getting
started guide, articles that explore specific use cases, and reference
pages for each function.
Bug reports, feature requests, and general questions can be submitted at https://github.com/cmmr/rbiom/issues.
Pull requests are welcome. Please ensure contributed code is covered by tests and documentation (add additional tests and documentation as needed) and that it passes all automated tests.
The following commands will check if rbiom passes the
bundled testing suite.
install.packages('testthat')
testthat::test_check('rbiom')These binaries (installable software) and packages are in development.
They may not be fully stable and should be used with caution. We make no claims about them.